
夏大豆重组自交系群体遗传图谱构建及开花期QTL 分析
2020-02-28 01:48:44曹永策李曙光张新草孔杰杰赵团结
中国农业科学 2020年4期
曹永策,李曙光,张新草,孔杰杰,赵团结
(1 延安大学生命科学学院/陕西省红枣重点实验室(延安大学),陕西延安 716000;2 南京农业大学大豆研究所/国家大豆改良中心/农业部大豆生物学与遗传育种重点实验室(综合)/作物遗传与种质创新国家重点实验室/江苏省现代作物生产协同创新中心,南京 210095)
0 引言
【研究意义】大豆是典型的短日照植物,对光温变化敏感,日照长度必须短于一定时长才能开花[1-2]。单一的大豆品种只能适应狭窄的纬度范围,在不同大豆产区都具有与之相适应的生态类型[3-4]。开花期是重要的生育期性状,不仅是大豆光温反应的直接体现,同时对其他的产量和品质性状具有重要影响[5-7]。因此,深入解析大豆开花期的遗传基础,对大豆品种的栽培区域合理划分以及培育广适应的大豆品种具有重要意义。【前人研究进展】大豆开花期是典型的数量性状,在遗传上既受主效基因控制,又受微效多基因影响[8]。目前,通过遗传分析报道了E1[9]、E2[9]、E3[10]、
E4[11]、E5[12]、E6[13]、E7[14]、E8[4]、E9[2]、E10[15]、J[16]
和E11[17]共12 个控制大豆生育期的基因。其中,E1[1]、E2[18]、E3[19]、E4[20]、E9[21]、E10[15]和J[22]位点的功能基因已被克隆或预测。E6还没有被定位,E5可能不是一个真实存在的位点[23]。除了这些被命名的基因位点外,还有许多数量控制位点(QTL)被报道[24-32]。目前,在SoyBase 数据库中至少报道了100 个控制大豆开花期的QTL(www.soybase.org)。另外KIM 等[33]通过同源比对分析发现在大豆中有118 个与拟南芥生育期相关的同源基因;同时,ZHAI 等[34]研究也发现即使E1—E4等开花期位点遗传背景相同,群体的开花期仍有分离。以上结果均表明仍有许多未知的QTL/基因控制开花期。【本研究切入点】上述研究使用的材料多为春大豆类型,而江淮地区是中国重要的大豆产区,品种以麦收后夏大豆类型为主,不同于国内外春大豆类型。但目前对该地区夏大豆开花期性状遗传基础的研究相对较少。【拟解决的关键问题】本研究以2 个夏大豆材料科丰35(KF35)和南农1138-2(NN1138-2)为亲本杂交衍生的1 个重组自交系群体(NJK3N-RIL)为试验材料,基于高密度遗传图谱,对大豆开花期性状进行QTL 分析,以揭示夏大豆开花期的遗传构成;同时鉴定出能在多个环境下稳定表达的QTL,为分子标记辅助选择育种和基因图位克隆提供有用信息。
1 材料与方法
1.1 试验材料与田间试验设计
以2 个夏大豆材料KF35 和NN1138-2 为亲本,杂交得到F1代,经单籽传法,衍生的91 个家系(F2:8)组成的重组自交系(RIL)群体(NJK3N-RIL)为试验材料。
NJK3N-RIL 群体以及2 个亲本在2012 年分别种植于南京农业大学农学试验站(2012JP)和安徽科技学院凤阳试验田(2012FY);在2013 年分别种植于南京农业大学农学试验站(2013JP)和安徽科技学院凤阳试验田(2013FY);在2014 年分别种植于南京农业大学农学试验站(2014JP)和江苏沿海地区农业科学研究所(2014YC),共计6 个生长环境。采用随机完全区组设计,3 次重复,单行区,行长1 m,每行10 个单株,株距10 cm,相邻行距50 cm,常规田间管理。
1.2 表型调查及数据分析
在单个种植重复中,NJK3N-RIL 群体及其亲本的开花期记录为从播种当日到试验小区有一半以上植株主茎出现花朵的天数,单个环境下表型值取该环境下3 个种植重复的平均值,记录单位为“天”[29,35]。
利用SPSS 20.0(SPSS Inc.,Chicago,IL,USA)软件对表型数据进行描述性统计,包括性状在环境下的变异范围、平均值、标准差、变异系数和正态分布检验等。使用SAS 9.3(SAS Institute Inc. 2011)软件的Proc GLM 过程进行多环境联合方差分析,估计方差分量用来评估性状在群体中的广义遗传率。广义遗传率计算公式如下:
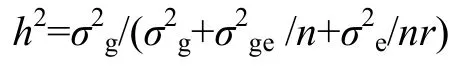
其中,σ2g为遗传方差、σ2ge为基因型与环境互作方差、σ2e为误差方差,n为环境数,r为单环境下的重复次数[36]。
1.3 基因型分型与遗传图谱构建
采用限制性酶切位点相关的DNA 测序(restriction- site Associated DNA sequencing,RAD-Seq)技术,对NJK3N-RIL 群体进行全基因组SNP 开发,然后根据亲本和家系的基因型开发bin标记用于构建遗传图谱,具体方法参考HAN 等[37]和HUANG 等[38]方法。采用JoinMap 4.0 软件构建遗传图谱,以标记间LOD 值大于3 进行连锁群划分,采用Regression mapping 算法和Kosambi 函数计算连锁群内标记的线性排列以及标记间的遗传距离(centiMorgan,cM)[39]。
1.4 QTL 分析
使用QTL Network 2.2 和Windows QTL Cartographer V2.5_011 对大豆开花期进行QTL 分析[40-41]。首先利用QTLNetwork 2.2 软件基于混合线性模型的复合区间作图法(MCIM)进行多环境下联合分析,以检测不同效应(加性效应、上位性、环境互作)对大豆开花期的影响。检验时设置参数如下:检测窗口大小及步长分别设置为10 和1 cM,排列测验(permutation test)1 000 次,以P=0.05 为统计检测阈值确定显著性QTL;其次利用Windows QTL Cartographer V2.5_011软件的复合区间作图法(CIM)进行单环境下QTL 定位及效应检测,以检测能在多环境中稳定表现的QTL。检验时设置参数如下:采用Zmapqtl 模型6(标准模型),窗口大小和步长分别设置为10 和1 cM,排列测验1 000 次,以P=0.05 为统计阈值,判断是否存在QTL。在不同环境或方法下,检测到的QTL 置信区间重叠,且加性效应方向一致,认为是同一个QTL。

表1 不同环境下NJK3N-RIL 群体及其亲本开花期(天)性状描述性统计Table 1 Results of descriptive statistics of flowering time (days) in NJK3N-RIL population grown in different environments
2 结果
2.1 NJK3N-RIL 群体开花期性状表型变异特点
NJK3N-RIL 群体及其亲本在多个种植环境下的开花期表型如表1所示。在所有环境下,2个亲本KF35和NN1138-2 的开花期平均值分别为40.4 和48.1 d,二者差异显著(P<0.01)。并且NJK3N-RIL 群体的开花期性状在单个环境中均呈连续分布;除2014JP环境外,其他5 个环境下该群体的开花期表型频率分布的偏度和峰度值(绝对值)均小于1,基本符合正态分布。表明大豆开花期性状为多位点控制的数量性状(表1 和图1)。
大豆开花期具有较高的广义遗传率(h2,93.8%),多环境联合方差分析结果表明(电子附表1),开花期性状在家系间、环境间以及环境与家系互作上均存在极显著差异(P<0.001),说明开花期在群体中存在较大的遗传变异,且易受环境影响。但家系与环境互作的均方值远小于家系间的均方值,说明了开花期性状在家系间的差异主要是由遗传变异引起的。
2.2 基因型分型与遗传图谱构建
采用RAD-seq 技术对NJK3N-RIL 群体及其亲本进行基因型分型。以Williams 82(Glyma.Wm82.a1. v1.1)为参考基因组,经过序列比对和过滤,共获得36 778 个高质量SNP 标记,分布在大豆20 条染色体上(表2)。其中只有第1、12 和19 染色体的SNP数低于1 000 个。根据窗口滑动法,将36 778 个SNP划分成1 733 个bin 标记用于遗传图谱构建和后续分析。
根据标记间的连锁情况,1 733 个bin 标记被划分为20 条连锁群,同大豆20 条染色体相对应。以连锁群为单位,计算标记的线性排列,并估算相邻标记间的遗传距离。最终构建出一张包含20 条连锁群,遗传长度为2 362.4 cM 的遗传图谱(表2、图2 和电子附图1)。标记间的平均距离为1.4 cM,平均每条连锁群的遗传长度为118.1 cM。其中,第1 染色体对应的连锁群上含有的bin 标记数目最少,为57 个;第18 染色体对应的连锁群上的bin 数目最多,为110个。第14 染色体所对应的连锁群遗传长度最短,为80.1 cM;第15 染色体所对应的连锁群遗传长度最长为148.6 cM。

图1 NJK3N-RIL 群体开花期在不同环境中的分布Fig. 1 Frequency distribution of flowering time in NJK3N-RIL population grown in different environments

表2 NJK3N-RIL 群体遗传图谱信息汇总Table 2 Summary of genetic map information of NJK3N-RIL population

图2 NJK3N-RIL 群体中bin 标记在遗传图谱上的分布Fig. 2 Distribution of bin markers on 20 Linkage groups in the NJK3N-RIL population

表3 NJK3N-RIL 群体开花期加性及QTL 与环境互作效应的分析Table 3 Additive QTLs and QTL-by-environment interaction effect for flowering time in the NJK3N-RIL population
2.3 利用MCIM 法检测开花期性状QTL
利用MCIM 法,在多环境联合分析下共检测到9个控制开花期的加性QTL,分布在第8、10、11、13、15、16 和20 染色体上(表3 和图3)。单个QTL 的加性效应值在-0.5—0.7 d,能够解释3.0%—12.0%的表型变异。其中,qFT-16-1和qFT-20-1的表型解释率均在10%以上,是该群体中主效QTL;qFT-11-1在2013FY 和2014JP 中与环境互作显著,互作效应值分别为0.6 和-0.8 d,互作效应对表型变异的总解释率为4.6%。在检测到的9 个加性QTL 中,除qFT-10-1外,其他所有 QTL 的增效等位变异来均来自亲本NN1138-2,这与NN1138-2 具有较晚的开花期相一致。
在NJK3N-RIL 群体中共检测到2 对上位性互作QTL,均为加性效应QTL 构成(表4 和图3)。其中,第一对上位性QTL 对由qFT-11-1和qFT-16-1构成,互作效应值和表型解释率分别为-0.2 d 和0.6%;并且该上位性互作QTL 对在2013FY 和2014JP 中与环境互作显著,能够解释0.8%表型变异。第二对上位性QTL 对由qFT-13-1和qFT-16-2构成,互作效应值和表型解释率分别为0.3 d 和1.5%,但与环境互作不显著。
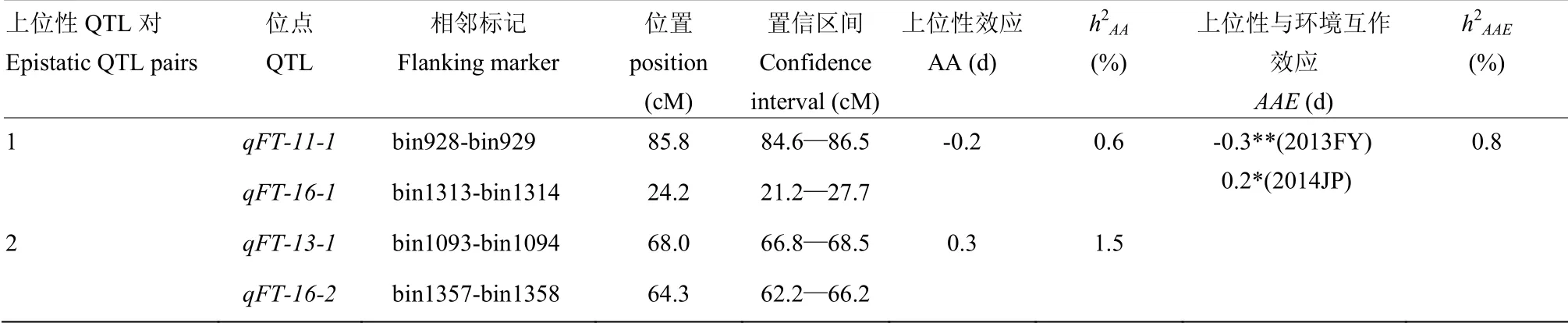
表4 NJK3N-RIL 群体开花期QTL 上位互作效应分析Table 4 Epistatic QTL pairs for flowering time in the NJK3N-RIL population
2.4 利用CIM 法检测开花期性状QTL
利用CIM 法,在NJK3N-RIL 群体中共检测到10个控制开花期的QTL,分布于第5、6、8、11、15、16和20染色体(表5和图3)。其中,qFT-8-1、qFT-11-1、qFT-15-1和qFT-16-1能够在3 个及以上的环境中检测到,是NJK3N-RIL 群体中控制开花期性状的环境稳定QTL。qFT-16-1能够在所有环境中检测到,单个环境中的LOD 值为4.7—13.9,解释了10.8%—28.9%的表型变异,是NJK3N-RIL 群体中的最稳定和主效QTL。qFT-11-1在5 个环境中检测到,单个环境下LOD 值为8.6—18.2,解释了17.7%—49.6%的表型变异,是NJK3N-RIL 群体中控制开花期性状另一个主效位点。qFT-15-1在4 个环境检测到,单个环境下LOD 值为4.0—9.3,解释了7.6%—23.0%的表型变异。qFT-8-1在3 个环境中检测到,单个环境下LOD 值在3.2—4.6,解释了6.6%—9.1%的表型变异。qFT-8-2在2 个环境中检测到,在单个环境中的LOD 值分别为3.8 和4.8,表型贡献率分别为5.9%和9.4%。qFT-5-1、qFT-6-1、qFT-16-2、qFT-20-1和qFT-20-2仅能在单环境中检测到,LOD 值为3.4—10.3,解释了5.0%—19.1%的表型变异,为环境敏感QTL。在检测到的10 个QTL 中,除qFT-5-1增效等位变异来自亲本KF35 以外,其余9个QTL 的增效等位变异均来自亲本NN1138-2。

图3 NJK3N-RIL 群体中开花期QTL 在连锁群上的位置Fig. 3 Locations of QTL for flowering time in soybean linkage map in NJK3N-RIL population

表5 NJK3N-RIL 群体不同环境中开花期QTL 分析Table 5 Detection of QTL associated with flowering time in the NJK3N-RIL population grown in different environments
3 讨论
3.1 bin 标记遗传图谱构建
QTL 定位是解析植物数量性状遗传基础的一种有效方法。遗传图谱的质量显著影响QTL 定位结果的精度,在特定群体中增加标记密度可以提高图谱的作图分辨率[42-43]。如ZHANG 等[44]应用一张包含6 159个SLAF 标记的大豆高遗传图谱分析耐低磷性状的QTL,发现同低密度遗传图谱相比,高密度遗传图谱的作图精度会显著提高。目前,下一代测序技术,具有高通量、标记开发周期短等特点,能够方便的、快速的获得全基因组范围内的SNP 标记,为高密度遗传图谱构建和数量性状解析提供了方便[45-47]。其中,RAD-seq 技术近年来被广泛应用于大豆遗传图谱构建、QTL 分析和候选基因挖掘等研究中。WANG 等[48]基于RAD-seq 技术构建了2 张分别包含5 728 和4 354个bin 标记的高遗传图谱,并对大豆花色、花期和种皮色等性状进行了分析,发现大部分的QTL 都被定位在小的基因组区间内;CHENG 等[49]利用该技术构建了一张包含3 469 个bin 标记的遗传图谱,将抗疫霉病位点RpsWY定位在约100 kb 区间内,并预测了4 个候选基因;WANG 等[50]利用RAD-seq 技术构建的图谱将叶型相关的一些QTL 定位在较小的基因组区间内,并对相关位点的候选基因进行分析。以上结果均表明基于RAD-seq技术构建的高密度遗传图谱具有较高的作图分辨率,能够方便地应用于QTL 定位和候选基因分析等研究中。而本研究利用该技术构建的图谱包含1 733 个bin 标记,标记间的平均距离为1.4 cM,可以保证定位到的QTL都有2 个或以上的紧密连锁标记(5 cM 内),能够在一定程度上弥补作图群体规模较小的缺陷,提高了作图分辨率。相应地,本研究的定位结果也显示了绝大多数QTL 被定位在1 Mb 的物理区间内。因此,本研究构建的遗传图谱能够有效地应用于大豆农艺、产量和品质等相关性状的遗传解析、分子标记辅助选择育种以及基因图位克隆的研究。
3.2 夏大豆NJK3N-RIL 群体开花期的遗传构成
本研究结果显示夏大豆NJK3N-RIL 群体开花期性状具有较高的遗传率(93.8%),但多环境联合方差分析结果也表明群体中材料的基因型差异、环境差异及基因型与环境互作效应均对开花期存在极显著影响,说明开花期受许多因素影响。XU 等[51]认为大部分数量性状的表型除受主效QTL 或基因控制外,还可能会受QTL 上位性和环境互作效应的影响。且当QTL作图模型中考虑了这些因素时,QTL 的定位精度会大大提高[52-53]。因此,本研究利用了具有加性、上位性及与环境互作效应的完整遗传模型进行QTL 分析。结果显示加性QTL、上位性互作QTL 及环境互作效应均对开花期表型变异有影响,多个QTL 存在环境互作或上位性互作效应,3 种效应累积表型变异解释率分别为63.9%、2.1%和4.6%。因此,如果在使用这些QTL 进行分子辅助育种时能够充分考虑这些遗传效应,将有利于更加准确地估计育种表型值。但在开花期遗传构成中,加性QTL 效应的贡献率要远大于另外2 种因素,占绝对优势,暗示了在前期研究中有必要去发掘能够在多个环境稳定表达的加性QTL。
3.3 与前人大豆开花期QTL 定位结果的比较
利用MCIM 法共检测到9 个控制开花期的加性QTL,利用CIM 法共检测到10 个控制开花期的QTL,且有7 个QTL 使用2 种方法均能检测到。综合2 种分析方法,本研究共检测到12 个开花期QTL。其中,qFT-6-1、qFT-10-1、qFT-11-1、qFT-16-1、qFT-16-2和qFT-20-1等分别与SoyBase 中报道的控制大豆开花期(first flower)位点First flower 1-1、First flower 24-4、First flower 11-2、First flower 13-7、First flower 9-3、First flower 21-2的区间相邻或重叠,推测可能是相同的QTL(表3 和表5),与前人研究相一致,从侧面验证了这些位点的真实性;同时也有 6 个位点(qFT-5-1、qFT-8-1、qFT-8-2、qFT-13-1、qFT-15-1和qFT-20-2)是本研究新检测到的开花期QTL(表3和表5),尤其是qFT-8-1和qFT-15-1能够在多个环境和不同方法中检测到,表明夏大豆NJK3N 群体中具有独特的开花期遗传基础构成。另外,本研究检测的QTL 与E1、E2、E3、E4和E9等一些在春大豆材料中控制开花期起重要作用的位点并不重合,表明江淮地区夏大豆开花期调控方式与春大豆的调控方式具有差异。因此,有必要利用更多的种质资源来揭示夏大豆开花期的遗传基础。
虽然在SoyBase 中有100 多个与大豆开花期相关的QTL 被报道,但大多数位点只能在单个或特定的环境中被检测到。鉴定出不同环境稳定表达的QTL,将方便应用于MAS 或基因图位克隆解析大豆开花期分子机制的研究。本研究中,qFT-8-1、qFT-11-1、qFT-15-1和qFT-16-1能够在3 个及以上环境和2 种方法同时检测到,是NJK3N-RIL 群体中控制开花期的环境稳定QTL(表3 和表5),能够作为分子辅助育种及基因图为克隆重要目标位点。
3.4 大豆开花期候选基因预测
植物开花的过程既受内在遗传机制控制,又受外界环境条件影响,调控机制复杂,有许多基因可能参与开花期的调控。但在大豆中只有少数几个开花期相关的基因被克隆和鉴定,发掘一些可能参与大豆开花调控的基因,能够充实对其分子调控机制的认识。本研究根据QTL 区间内基因功能的注释及相关文献,对4 个环境稳定QTL 的候选基因进行了预测。qFT-8-1区间内发现2 个AP2 亚族转录因子(Glyma08g38190和Glyma08g38800),前人研究结果显示AP2 亚族转录因子对花的增殖生长至关重要[54]。qFT-11-1区间内发现Glyma11g18940和Glyma11g18290参与光周期调节和花形态建成(www.soybase.org)。qFT-15-1区间内有 3 个基因最有可能参与调控大豆开花期,Glyma15g41740编码一个MYB 家族蛋白,具有序列特异性DNA 结合转录因子的活性,可以激活FT 的互作蛋白FTIP1,实现促进开花[55];Glyma15g41770编码一个未知功能的蛋白,但该基因可能参与花发育的生物过程(http://www.Soybase.org);Glyma15g41825编码一个NAM 蛋白,是植物特异性转录因子,前人研究结果表明NAM 结构域蛋白在植物发育过程中发挥着重要作用,包括参与顶端分生组织发育、花的形态建成和胁迫诱导开花响应等[56-57]。qFT-16-1区间内包含拟南芥同源基因GmFT5a,KONG 等[58]研究表明GmFT5a调控大豆光周期反应。但这些区间内的真正功能基因还需要通过分子生物学等手段进一步验证。总的来讲,这些位点及其紧密相关的分子标记为大豆分子育种及基因图为克隆提供了重要的信息。
4 结论
夏大豆开花期遗传构成复杂,加性QTL 效应占绝对优势,上位性互作及环境互作效应对开花期影响较小。共检测到12 个控制开花期的QTL,其中4 个QTL(qFT-8-1、qFT-11-1、qFT-15-1和qFT-16-1)能够在多个环境和使用不同分析方法检测到,是NJK3N-RIL群体中控制开花期重要位点。
猜你喜欢
中国农学通报(2022年29期)2022-11-25 07:28:12
少先队活动(2020年12期)2021-01-14 01:47:40
现代园艺(2017年21期)2018-01-03 06:41:32
中成药(2017年3期)2017-05-17 06:09:01
Acta Mathematica Scientia(English Series)(2016年5期)2016-11-24 11:59:31
甘肃林业科技(2016年2期)2016-11-16 09:15:39
领导科学论坛(2016年9期)2016-06-05 14:59:58
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
