
广金钱草多糖指纹图谱的建立、含量测定及其对α-葡萄糖苷酶的抑制活性研究
2020-02-24 09:10程轩轩陈亮元郑诗嘉唐晓敏杨全
中国药房 2020年2期
程轩轩 陈亮元 郑诗嘉 唐晓敏 杨全
中图分类号R284.1
文献标志码A
文章编号1001-0408(2020)02-0183-07
DOI
10.6039/j .issn.1001-0408.2 02 0.02.11
摘要 目的:建立广金钱草多糖的指纹图譜,分析其单糖组成及含量,并考察广金钱草多糖对α一葡萄糖苷酶的体外抑制作用。方法:采用水提醇沉法提取广金钱草多糖,经三氟乙酸水解、1-苯基-3-甲基-5-吡唑啉酮衍生化后,采用高效液相色谱法(HPIC)建立指纹图谱(以葡萄糖峰为参照),并进行单糖组成及含量分析。含量测定色谱柱为Phenomenex LunaC18,流动相为乙腈-0.05mol/L磷酸二氢钾溶液(用氢氧化钠调节pH至6.8),梯度洗脱,流速为0.8 mL/min,检测波长为250 nm,柱温为30℃,进样量为10μL。以阿卡波糖为对照,采用对硝基苯-a-D-葡萄糖吡喃苷法考察广金钱草多糖对α一葡萄糖苷酶的体外抑制活性。结果:18批药材样品的HPLC指纹图谱有9个共有峰,其中15批药材样品的相似度大于0.90;共指认甘露糖、鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、木糖、阿拉伯糖等7个共有峰。其中,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖的含量分别为0.471- 2.092、1.379-8.919、2.560-35.679、1.194- 6.905、0.566-4.158 mg/g;以鼠李糖为基准,其余4种单糖的摩尔比分别为1.58-4.07、2.26-19.95、2.20-4.21、1.31-2.86。广金钱草多糖对α一葡萄糖苷酶的抑制活性呈现随剂量升高而增强的趋势,其半数抑制浓度为0.70 mg/mL,低于阳性对照阿卡波糖(7.76 mg/mL)。结论:不同批次广金钱草的多糖组成均以葡萄糖为主,各单糖含量有所差异。广金钱草多糖对α一葡萄糖苷酶具有明显的体外抑制作用,且活性强于阿卡波糖。
关键词 广金钱草多糖;单糖;含量;指纹图谱;高效液相色谱法;α一葡萄糖苷酶;抑制活性步研究证实。
广金钱草为豆科植物广金钱草[Desmodium styraci-folium (Osb.) Merr.]的干燥地上部分,味甘、淡,性凉,归肝、肾、膀胱经,具有利湿退黄、利尿通淋之效,主治黄疸尿赤、热淋、石淋、小便涩痛等症[1]。广金钱草中的黄酮类成分因具有利尿、抗氧化、抑制大鼠肾结石形成、改善大鼠离体心脏缺血再灌注损伤等广泛的药理活性,是评价药材质量的指标性成分,备受学者关注[2-5]。除黄酮类成分外,多糖在广金钱草中的含量也较高,但其相关药理学研究较少。考虑到植物多糖在抗肿瘤[6]、抗衰老[7]、免疫调节[8]、降血脂[9]等方面独特的生物活性,开发前景良好,故本研究拟从广金钱草药材中提取多糖,采用1一苯基一3一甲基-5一吡唑啉酮(PMP)柱前衍生化一高效液相色谱法(HPIC)分析其单糖组成,并建立不同产地广金钱草多糖的指纹图谱;在测定多糖含量的基础上,初步评价其对α一葡萄糖苷酶的抑制作用,旨在为全面开发利用该药材资源提供依据。
l 材料
1.1 仪器
LC-20AT型HPLC仪,配有SPD-M20A型二极管阵列(DAD)检测器、SIL-20A型自动进样器、CT0-20A型柱温箱、DGU-20A3型真空脱气机、LC-20AT型溶液传输单元(日本Shimadzu公司);BP211D型十万分之一电子天平(德国Sartorius公司);TG-16WS型台式高速离心机(长沙湘智离心机仪器有限公司);UV-5200PC型紫外一可见分光光度计(上海元析仪器有限公司);Multi-skan GO型酶标仪(美国Thermo Fisher Scientific公司);Biosafer-20TB型实验室级超纯水器[赛飞(中国)有限公司];XMTD-8222型电热恒温水浴锅、PH27-9240A型电热恒温鼓风干燥箱(上海精宏实验设备有限公司);ST2100型实验室pH计[奥豪斯仪器(常州)有限公司];N-1100D-WD型旋转蒸发仪(日本Eyela公司)。
1.2 药品与试剂
葡萄糖对照品(批号:AF7022401)、阿拉伯糖对照品(批号:AF8051683)均购自成都埃法生物科技有限公司;甘露糖对照品(批号:40708)、半乳糖对照品(批号:10509)、鼠李糖对照品(批号:30521)均购自德国Dr. Eh-renstorfer GmbH公司;半乳糖醛酸对照品(上海金穗生物科技有限公司,批号:20180422);木糖对照品(上海麦克林生化科技有限公司,批号:C10026034);α一葡萄糖苷酶对照品(美国Sigma公司,批号:SLBX6245);阿卡波糖对照品(批号:J1824027)、对硝基苯-a-D-葡萄糖吡喃苷( PNPG)对照品(批号:E1828029)均购自阿拉丁试剂(上海)有限公司;上述对照品的纯度均大于98%。0.1mol/L磷酸盐缓冲液(PBS) (pH 6.8,福州飞净生物科技有限公司);乙腈、磷酸二氢钾(KH2P04)、PMP等均为色谱纯,其余试剂均为分析纯,水为超纯水。
1.3 药材
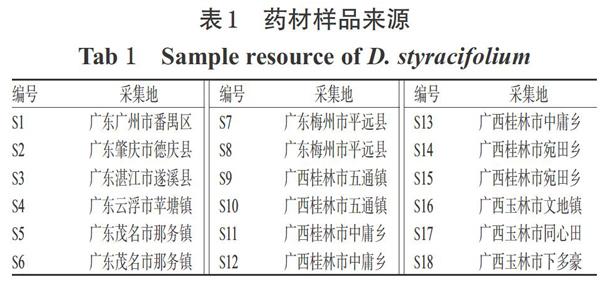
广金钱草药材(编号:SI~S18)采自广东省各栽培基地,经广东药科大学中药学院中药资源系杨全教授鉴定为豆科植物广金钱草[D.styracifolium( Osb.) Merr.]的地上部分。药材样品来源见表1。
2 方法与结果
2.1 色谱条件
色谱柱:Phenomenex Luna C18( 250 mm×4.6 mm,5μm);流动相:乙腈(A相)-0.05 mol/L KH2PO4溶液(用氢氧化钠调节pH至6.8,B相),梯度洗脱(0~30 min,17%A→ 19%A;30~50 min,19%A);流速:0.8 mL/min;检测波长:250 nm;柱温:30℃;进样量:10 μL。
2.2 溶液的制备
2.2.1 混合单糖对照品贮备液及其衍生化溶液
精密称取鼠李糖、半乳糖醛酸、葡萄糖、半乳糖和阿拉伯糖对照品各适量,置同一量瓶中,加水配制成质量浓度分别为0.33、0.88、1.89、0.82、0.45 mg/mL的混合单糖对照品贮备液。精密吸取上述混合單糖对照品贮备液200 μL,置于具塞试管中,依次加入0.3 mol/L氢氧化钠溶液200 μL和0.5 mol/L PMP甲醇溶液200 μL,混匀,密封,于70℃反应1h,取出,冷却至室温;精密加入0.3mol/L盐酸溶液200 μL调pH至中性,加入12.5%KH2PO4溶液200 μL,摇匀,再加入等体积氯仿萃取3次;弃去氯仿液,上清液经0.22 μm微孔滤膜滤过,即得混合单糖对照品衍生化溶液,备用。
2.2.2 粗多糖及其供试品溶液
采用水提醇沉法提取广金钱草多糖。称取广金钱草药材样品适量,粉碎,取粉末5 9,用沸水200 mL回流提取3次,每次1h;趁热滤过,合并滤液,浓缩后以5 000r/min离心10 min;取上清液,加适量乙醇至含醇量为80%(V/V),于4℃冰箱中放置过夜;以5 000 r/min离心5 min,收集沉淀,冻干,即得粗多糖。精密称取上述粗多糖50 mg,置具塞试管中,加入2 mol/L三氟乙酸溶液2 mL,混匀,密封,于100℃水解8h,蒸干,残渣加甲醇1mL,蒸干,重复3~5次;残渣加水8 mL复溶,即得多糖水解液。取上述多糖水解液200 μL置于具塞试管中,按“2.2.1”项下方法进行衍生化处理,即得供试品溶液,备用。
2.2.3 阴性对照溶液
按“2.2.1”项下方法制备不含单糖对照品的阴性对照溶液,备用。
2.3 HPLC指纹图谱的建立
2.3.1 方法学考察
(l)精密度试验:取“2.2.2”项下供试品溶液(编号:S16)适量,按“2.1”项下色谱条件连续进样6次,以葡萄糖峰为参照,记录各共有峰的相对保留时间和相对峰面积。结果,9个共有峰相对保留时间和相对峰面积的RSD均小于3%(n=6),表明该方法的精密度良好。
(2)稳定性试验:取“2.2.2”项下供试品溶液(编号:S16)适量,分别于室温下放置O、4、8、12、16、24 h时按“2.1”项下色谱条件进样测定,以葡萄糖峰为参照,记录各共有峰的相对保留时间和相对峰面积。结果,9个共有峰相对保留时间和相对峰面积的RSD均小于5%(n=6),表明供试品溶液于室温放置24 h内基本稳定。
(3)重复性试验:取药材样品(编号:S16)适量,粉碎,精密称取粉末适量,共6份,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,以葡萄糖峰为参照,记录各共有峰的相对保留时间和相对峰面积。结果,9个共有峰相对保留时间和相对峰面积的RSD均小于5%(n=6),表明本方法重复性良好。
2.3.2 HPLC指纹图谱生成与相似度、共有峰相关分析
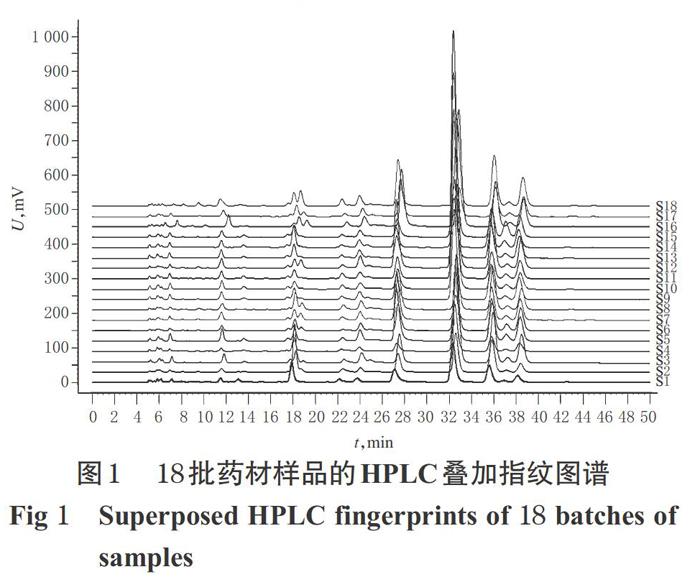
(l) HPLC指纹图谱的生成:取18批药材样品各适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定。采用《中药色谱指纹图谱相似度评价系统》(2012版)对18批药材样品的HPLC指纹图谱进行分析,得HPLC指纹图谱,详见图1、图2。
(2)相似度评价:采用《中药色谱指纹图谱相似度评价系统》(2012版),以药材样品的HPLC对照指纹图谱为对照,进行整体相似度评价。结果显示,18批药材样品中,有15批相似度大于0.90,详见表2。
(3)共有峰的指认及相关分析:18批药材样品有9个共有峰,通过与混合单糖对照品衍生化溶液(按“2.2.1”项下方法配制,甘露糖、鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、木糖、阿拉伯糖的质量浓度分别为0.24、0.13、0.35、0.76、0.33、0.07、0.18 mg/mL,其HPLC图见图3A)比对,指认出1、4、5~9号峰依次为甘露糖、鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、木糖、阿拉伯糖。由于6号峰信号强且峰形好,故以其保留时间和峰面积为参照,计算其他色谱峰的相对保留时间和相对峰面积,详见表3、表4(表中,P1~P9依次对应1~9号峰)。
2.4 广金钱草多糖的单糖含量测定
由于7个共有峰中,有2个单糖(甘露糖、木糖)的含量偏低(低于本方法检测限),故本研究只检测了其余5个单糖的含量。
2.4.1 方法学考察
(l)专属性:取“2.2”项下混合单糖对照品衍生化溶液、供试品溶液和阴性对照溶液各适量,按“2.1”项下色谱条件进样测定,记录色谱图。结果,对照品和供试品溶液的色谱峰峰形良好,阴性对照溶液对测定无干扰,详见图3。
(2)线性关系考察:精密吸取“2.2.1”项下混合对照品贮备液0.05、0.1、0.2、0.4、0.8、1.2、1.6、2.O mL,分别置于2 mL量瓶中,加水稀释至刻度,混匀。再分别精密吸取上述稀释后的混合对照品溶液各200 μL,按“2.2.1”项下方法进行衍生化处理,再按“2.1”项下色谱条件进样测定。以待测物质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行线性回归,得鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖的回归方程分别为y=13 534x - 63 714 (r=0.999 8)、y=17 053x - 94 675 (r=0.999 6)、y=16 639x+25 588( r=0.999 4)、y=20 818x-9 100.9 (r=0.999 7)、y=20 065x-42 258(r=0.999 6).上述各单糖检测质量浓度的线性范围分别为8.25~330.00、22.00~880.00、47.25~1 890.00、20.50~820.00、11.25~450.00 μg/mL。
(3)检测限与定量限考察:精密吸取“2.2.1”项下混合单糖对照品衍生化溶液适量,以水为溶剂倍比稀释,并按“2.1”项下色谱条件进样测定,分别以信噪比3:1和10:1计算检测限和定量限。结果,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖的检测限分别为2.06、2.75、2.36、1.03、2.25 μg/mL;定量限分别为4.13、11.00、5.91、2.56、5.63 μg/mL。
(4)精密度试验:精密吸取“2.2.1”项下混合单糖对照品衍生化溶液适量,按“2.1”项下色谱条件连续进样6次,记录峰面积。结果,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖峰面积的RSD分别为1.48%、3.49%、0.33%、0.64%、0.41%(n=6),表明仪器精密度良好。
(5)稳定性试验:取“2.2.2”项下供试品溶液(编号:S16)适量,分别于室温下放置0、2、4、8、12、24 h时按“2.1”项下色谱条件进样测定,记录峰面积。结果,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖峰面积的RSD分别为1.53%、2.99%、3.68%、5.82%、4.99%(n=6),表明供试品溶液于室温放置24 h内基本稳定。
(6)重复性试验:取药材样品(编号:S16),共6份,粉碎,精密称取粉末适量,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件進样测定,记录峰面积并按标准曲线法计算样品含量。结果,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖含量的RSD分别为4.53%、4.72%、4.25%、4.71%、4.43%(n=6),表明该方法重复性良好。
(7)加样回收率试验:精密称取已知含量的样品(编号:S16)6份,每份50 mg,分别精密加入鼠李糖100μg、半乳糖醛酸240 μg、葡萄糖500μg、半乳糖240 μg、阿拉伯糖150 μg,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果,鼠李糖、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖的平均加样回收率分别为100.77%、102.61%、98.78%、100.60%.101.93% ,RSD分别为3.18%、2.08%、1.70%、1.83%、2.61%(n=6),表明该方法的准确度良好。
2.4.2 单糖含量测定与摩尔比计算
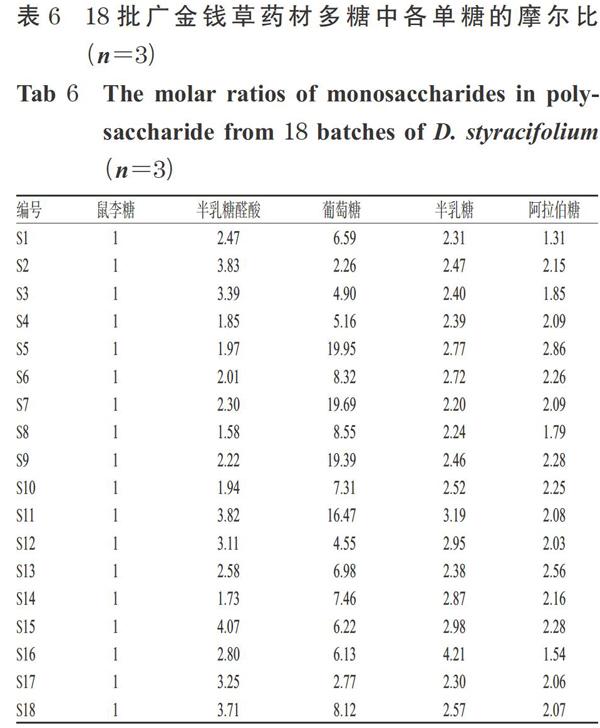
分别取18批广金钱草药材各适量,粉碎,精密称取药材粉末5 g,按“2.2.2”项下方法制备供试品溶液,再按“2.1”项下色谱条件进样测定。采用标准曲线法计算样品含量[以每克药材中所含单糖质量(mg)计];参考文献方法[10],以含量最低的单糖为基准,计算各单糖的摩尔比。结果,不同批次广金钱草药材中的多糖均以半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖为主,鼠李糖含量均最低,详见表5、表6。
2.5 广金钱草多糖对α一葡萄糖苷酶的体外抑制活性采用PNPG法进行考察。
2.5.1阿卡波糖对照品溶液的制备
精密称取阿卡波糖对照品适量,加水溶解并稀释,制得质量浓度分别为2.50、5.00、7.50、10.00、15.00、20.00、25.00 mg/mL的对照品系列溶液。
2.5.2 多糖供试品溶液的制备
精密称取“2.2.2”项下广金钱草粗多糖20 mg,加水400 μL使溶解,得粗多糖贮备液。分别量取上述贮备液各适量,加水稀释,制得质量浓度分别为0.50、1.00、1.50、2.00、2.50、4.00 mg/mL的多糖供试品溶液。
2.5.3 测定方法及结果
精密吸取“2.5.1”“2.5.2”项下的阿卡波糖对照品系列溶液和供试品溶液各10 μL,置于96孔板中,加入PBS 60 μL,再加入2U/mLα一葡萄糖苷酶水溶液10 μL,混匀,于37℃孵育15 min;随后加入5 mmol/L PNPG水溶液20 μL,于37℃孵育20 min,加入0.1 mol/L碳酸钠溶液50 μL终止反应。以水为空白对照,以阿卡波糖为阳性对照,于405 nm波长下测定各孔光密度(OD)值,计算抑制率和半数抑制浓度(ICso):抑制率(%)=(OD空n - OD 样品)/OD空白×100%[11]。各样品均重复测定3次,结果见表7。由表7可知,阿卡波糖和广金钱草多糖对α一葡萄糖苷酶的抑制活性均呈现随着剂量升高而增强的趋势;广金钱草多糖对α一葡萄糖苷酶抑制活性明显(IC50=0.70 mg/mL),且强于阳性对照阿卡波糖(IC50=7.76 mg/mL)。
3 讨论
3.1 衍生化条件的优化 本课题组前期以单糖峰面积为评价指标,分别对广金钱草多糖的酸水解时间、PMP衍生化温度及衍生化时间进行优化,得到最佳的柱前衍生化条件(即酸水解8h,70℃下衍生化1 h)。在PMP衍生化过程中,本课题组还比较了磷酸盐( KH2P04)的加入对样品HPLC色谱图的影响。结果发现,于氯仿萃取前在供试品液中加入适量KH2P04溶液,可明显抑制PMP峰,改善基线状态,有利于单糖的定量分析。
3.2 单糖检测方法的选择
生物多糖中单糖组成分析常用的方法包括薄层色谱法、气相色谱峰、HPLC法等。其中,薄层色谱法样品处理繁琐,鉴别能力弱,已逐渐被仪器分析方法取代;气相色谱法对中性单糖的分离效果较好,而对酸性多糖的分离效果较差,且前期处理复杂、耗时,易出现色谱峰异构化裂分;HPLC法可较好地对中性、酸性多糖进行分离鉴定[12]。故本研究采用PMP柱前衍生化-HPLC法对广金钱草多糖的单糖组成进行定性、定量分析。结果显示,所建HPLC法重复性、稳定性良好,且灵敏度较高。
3.3 色谱条件的优化
本课题组分别比较了乙腈一乙酸铵溶液和乙腈一KH2P04溶液等两种流动相体系,发现后者的分离效果更优。进一步对KH2P04溶液的浓度(0.01、0.02、0.05mol/L)进行筛选后发现,以0.05 mol/L KH2P04溶液(用氢氧化钠调节pH至6.8)作为有机相时,各待测物色谱峰峰形的对称性良好。此外,本课题组还考察了各单糖在YMC-Pack ODS-AQ、Inertsil ODS-3、Phenomenex Lu-na C18等色谱柱上的分离效果,最终确定了本文中的色谱条件。
3.4 指纹图谱的结果分析
本研究建立了18批广金钱草药材多糖的HPLC指纹图谱,确定了9个共有峰,并指认了其中7个。18批样品中有15批的相似度超过0.9,表明广金钱草多糖具有较好的稳定性,可作为药材质量控制的指标之一。
3.5 单糖定量分析结果分析
本课题组在18批广金钱草药材多糖中共检测了5种单糖成分的含量,分别为鼠李糖0.471~2.092 mg/g,半乳糖醛酸1.379~8.919 mg/g,葡萄糖2.560~35.679mg/g,半乳糖1.194~6.905 mg/g,阿拉伯糖0.566~4.158 mg/g;以鼠李糖为基准,半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖的摩尔比分别为1.58~4.07、2.26~19.95、2.20~4.21、1.31~2.86。结果表明,不同产地广金钱草的多糖组成均以葡萄糖为主,鼠李糖的含量最低,且各单糖含量存在差异,可能与产地的土壤、气候等环境因素有关。
3.6 α一葡萄糖苷酶抑制活性的结果分析
PNPG经α一葡萄糖苷酶催化可生成对硝基苯酚(PNP),PNP在400 nm左右波长处有最大紫外吸收,测定此波长下PNP的OD值,可用以计算抑制剂对α一葡萄糖苷酶的抑制率[13]。基于此,本研究以PNPG為底物、以阿卡波糖为阳性对照,初步评价了广金钱草多糖对α一葡萄糖苷酶的抑制作用。结果发现,广金钱草多糖对α一葡萄糖苷酶具有明显的体外抑制作用,且活性强于阳性对照阿卡波糖,可为后续其降血糖作用研究提供依据。
4 结语
本研究采用PMP柱前衍生化-HPLC法分析了18批广金钱草药材多糖中的单糖组分,并建立了广金钱草多糖的HPLC指纹图谱;在此基础上,通过对单糖组分的定性、定量分析,初步探讨了18批广金钱草药材多糖的组成特征、单糖含量及摩尔比。本研究还发现,广金钱草多糖对仅一葡萄糖苷酶的体外抑制活性强于阳性对照阿卡波糖,提示其可能具有降血糖作用。但本研究尚未对广金钱草多糖的药效学进行分析,有待后续研究进一步完善。
参考文献
[1]国家药典委员会,中华人民共和国药典:一部[S].2015年版.北京:中国医药科技出版社,2015:44.
[2]王鑫,广金钱草中黄酮类化合物定性分析及其抗大鼠肾草酸钙结石作用的研究[D].石家庄:河北医科大学, 2018.
[3]
MI J, DUAN JM, ZHANG J, et al.Evaluation of antiuro-lithic effect and the possible mechanisms of Desmodiumstyracifolium and Pyrrosiae petiolosa in rats[J].Urol Res,2012. 40(2):151-161.
[4]李冠烈,汤少娴,饶智华,等,广金钱草总黄酮对大鼠离体心脏缺血再灌注损伤的分子保护作用研究[J].生物化工,2018,4(3):97-101.
[5]
ZHOU C,LUO JG, KONG LY. Quality evaluation of Des-modium styracifolium using high-performance liquid chro-matography with photodiode array detection and electro-spray ionization tandem mass spectrometry[J]. PhytochemAnal,2012,23(3):240-247.
[6]苏畅,李小江,贾英杰,等.香菇多糖的抗肿瘤作用机制研究进展[J].中草药,2019,50(6):1499-1504.
[7]李海春.8种石斛多糖抗氧化、抗衰老活性的比较研究[D].广州:华南农业大学,2016.
[8]王娅,闫丽娜,孙甜甜,等.何首乌多糖的结构表征及其免疫调节活性研究[J].中草药,2019,50(10):2290-2295.
[9]杨夏,冯颖淑,童珊珊,等.降血脂多糖活性机制及构效关系研究进展[J].中国中药杂志,2018,43(20):4011-4018.
[10]陈思.茶枝柑多糖提取、分离纯化、结构及抗氧化活性研究[D].广州:广东药科大学,2011.
[11]阿卜杜热合曼·努如拉,阿米乃木·买买提,艾再提·克热木,等,榅椁子不同提取部位对α一葡萄糖苷酶的抑制活性及其降血糖作用[J].食品安全质量检测学报,2018,9(15):4018-4023.
[12]王贵春,陈雪峰,刘宁,等,柱前衍生化高效液相色谱法分析发菜胞外多糖的单糖组成[J].食品工业科技,2015,36(10):59-61.
[13]朱海雯,藏药绿萝花的化学成分和α一葡萄糖苷酶抑制活性研究[D].重庆:重庆大学,2015.
(收稿日期:2019-05-24修回日期:2019-10-18)
(编辑:张元媛)
△基金项目:国家发展和改革委员会新兴产业重大工程包/国家中医药管理局中药标准化行动计划项目(No.ZYBZ-Y-GD-13/ZYY-2017-099)
*副教授,博士。研究方向:中药化学成分与药理活性。电话:020-39352176. E-mail: gdyxycxx@126.com
#通信作者:教授,博士。研究方向:道地药材质量及形成机制。电话:020-39352353.
E-mail: yangquan7208@vip.163.com
猜你喜欢
中国药学药品知识仓库(2022年8期)2022-05-09
好日子(2020年6期)2020-07-09
中国中药杂志(2016年22期)2017-02-13
中国民族民间医药·上半月(2016年12期)2017-01-11
中国医药导报(2016年30期)2016-12-28
中国民族民间医药·上半月(2016年11期)2016-12-26
中国中药杂志(2016年20期)2016-11-19
科技资讯(2015年19期)2015-10-09
天津农业科学(2015年5期)2015-05-30
中国民族民间医药·下半月(2014年4期)2014-09-26
