
Toll样受体9基因多态性和幽门螺杆菌感染的关系
2020-02-14 10:30:30卫星如马立聪田旭阳贾彦彬
安徽医科大学学报 2020年1期
卫星如,马立聪,,田旭阳,党 彤,高 芳,贾彦彬,,4
幽门螺杆菌(helicobacterpylori,H.pylori)是革兰阴性细菌,感染了全球50%左右的人口[1]。H.pylori感染与胃肠道疾病的发生发展有关[2]。然而关于其作用机制尚不明确,影响H.pylori感染的因素也不明确。环境和宿主等多种因素的综合作用引起了H.pylori的感染,其中宿主因素起着很重要的作用[3],而且宿主的遗传改变可以影响H.pylori的感染状况及临床转归[4]。研究[5]发现,Toll样受体(Toll-like receptors, TLRs)作为一种重要的模式受体可以识别多种病原体关联的分子模式,进而触发信号传导的级联反应,致使多种炎性因子的生成,其对于识别H.pylori感染以及刺激机体产生免疫应答极其重要。该研究旨在检测TLR9基因单核苷酸多态性(single nucleotide polymorphism, SNP)与H.pylori感染之间的关系,为以TLRs为基础的抗H.pylori感染治疗提供一定的理论依据。
1 材料与方法
1.1 病例资料收集683例正常体检者,其中男性459例,女性224例。所有研究样本均为在包头生活5年以上,互无血缘关系的汉族人。所有入选者均无癌症病史,无高血压、糖尿病,未接受过H.pylori清除治疗及胃部手术,并通过内镜窄带成像术(narrow band imaging, NBI)胃镜检查排除了萎缩性胃炎、不完全肠转化、胃及十二指肠溃疡等疾病患者,所用样本采集血样2 ml,并提供了知情同意书,本研究经包头医学院医学伦理学委员会批准。
1.2H.pylori的检测用酶联免疫吸附测定方法(enzyme-linked immunosorbent assay, ELISA)检测样本中抗H.pylori的特异性抗体,人幽门螺旋杆菌抗体(helicobacter pylori antibody, HP-Ab)ELISA检测试剂盒购自泉州市睿信生物科技有限公司,操作及结果判断严格按照说明书进行。
1.3 SNP的筛选根据Hap Map数据库(http://www.Hapmap.org)所提供的中国汉族人群的信息,使用Agorithm-Tagger-pairwise Tagging软件筛选标记单核苷酸多态性(tag-SNP),要求最小等位基因频率(minimum allele frequency, MAF) ≥5%,连锁不平衡值r2>0.8。在筛选出的SNP中,选择文献报道可能和疾病关联的SNP进行研究,本研究选择了3个SNP,即rs187084、rs352140和rs164640进行研究。
1.4 基因分型采用DNA提取试剂盒(天根生物科技有限公司)提取外周血白细胞DNA。采用聚合酶链式反应-限制性片段长度多态性(polymerase chain reaction-restriction fragement length polymorphism,PCR-RFLP)技术对TLR9 rs187084、rs352140、rs164640进行基因分型。用Primer 3设计引物,由上海捷瑞生物工程有限公司合成引物。PCR反应体系共25 μl,内含DNA 50 ng、PCR mix 12.5 μl、上下游引物(5 μmol/L)各1 μl。PCR反应条件:94 ℃预变性5 min,94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸60 s,32个循环,最后72 ℃延伸5 min。PCR引物序列及产物长度见表1。扩增产物用相应的限制性内切酶进行酶切,酶切反应体系共10 μl,内含PCR反应产物3 μl,10×Buffer 1 μl,内切酶3 U。在内切酶最适反应温度下作用60 min后,2%琼脂糖凝胶电泳,根据酶切割后出现的片段大小判断基因型,内切酶及相应的反应温度和酶切片段长度见表1。
1.5 统计学处理采用SPSS 21.0软件进行统计分析。采用检验检测样本是否符合Hardy-Weinberg平衡(hardy-weinberg equilibrium, HWE)。采用Haploview 4.0软件对SNP位点进行连锁不平衡(linkage disequilibrium, LD)分析,用D’置信区间法构建单体型块(Haplotype block)。采用非条件Logistic回归检测各SNP在共显性(codominant)、显性(dominant)、隐性(recessive)遗传模型下与H.pylori感染的关系,以比值比(odds ratio,OR)值以及95%置信区间(confidence intervals,CI)表示,并计算最小信息准则(an information criterion, AIC)、贝叶斯信息准则(bayesian information criterion, BIC)。其中,AIC、BIC是对模型拟合效果进行评价的指标,AIC、BIC越小,则模型对数据的拟合越好,模型则为最佳模型。
2 结果
2.1 一般临床资料比较在683例样本中,H.pylori阴性305例,年龄39~88(58.71±10.86)岁;男性206例,占67.54%,女性99例,占32.46%。H.pylori阳性378例,年龄38~89(58.77±11.36)岁;男性253例,占66.93%,女性125例,占33.07%。研究对象的年龄和性别分布在H.pylori阴性组和阳性组间的差异无统计学意义(年龄:t=0.072,P=0.365; 性别:t=0.028,P=0.866)。
2.2 TLR9基因SNP位点与H.pylori感染的关系在683例样本中,TLR9基因rs187084、rs352140和rs164640位点的基因型分布均符合Hardy-Wein-berg平衡定律。分别用3种遗传模式对TLR9与H.pylori感染的关系进行分析。SNP rs187084在codominant、dominant、recessive模型中均与H.pylori感染关联,其中codominant模型为最佳模型,与TT基因型相比,携带TC基因型可使H.pylori感染的风险降低(TCvsTT:OR=0.520, 95%CI=0.329~0.820)。rs352140在3种遗传模式下,均未发现其与H.pylori感染具有相关性。rs164640在dominant模型中,与野生型AA基因型相比,携带AG+GG基因型者H.pylori感染风险增加1.569倍(95%CI=1.126~2.187)。见表2。对TLR9基因的3个SNP进行连锁不平衡分析发现:rs164640、rs352140、rs187084三个位点连锁程度低,未能构建出单体型块。
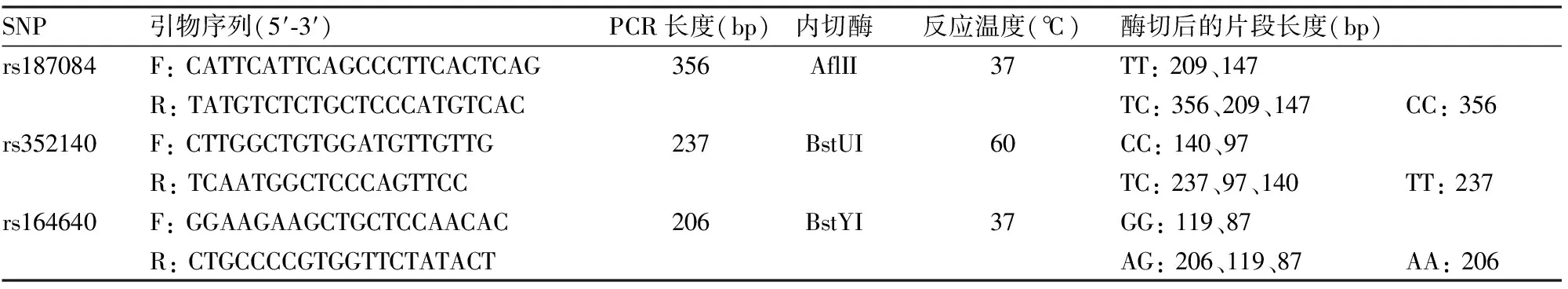
表1 TLR9 rs187084、rs352140和rs164640引物序列、内切酶和反应温度及酶切片段长度
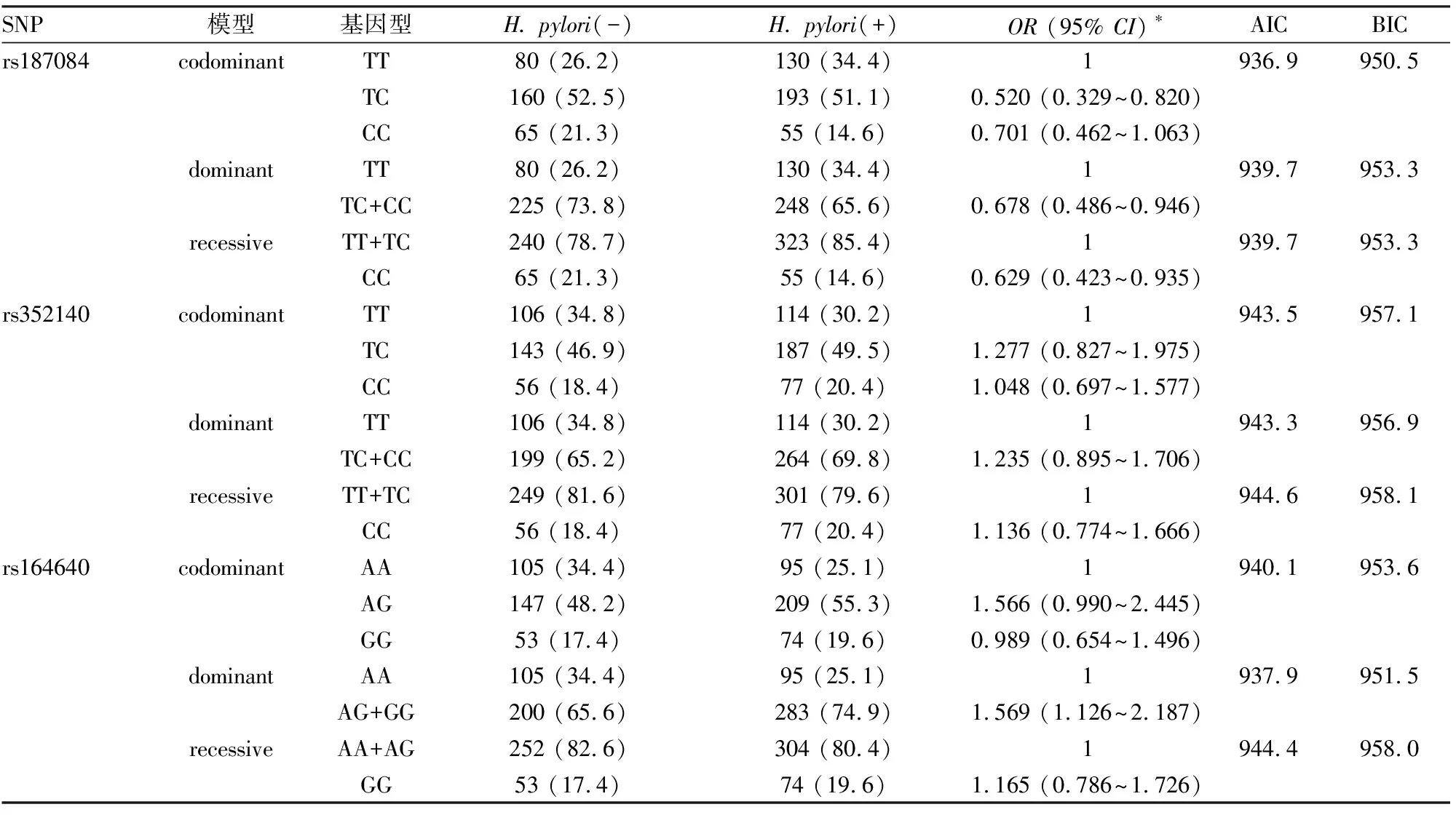
表2 不同遗传模式下,TLR9基因SNP位点与H. pylori感染风险的关系[n(%)]
*调整了年龄和性别因素
3 讨论
病原微生物侵入人体时受到模式识别受体的识别,进而触发信号传导的级联反应,使机体产生免疫应答。TLRs特异性地识别多种病原体关联的分子模式,在机体的非特异性免疫应答和承载特异性免疫应答中起到了重要作用[5]。研究[6]表明TLRs基因多态性与H.pylori的感染有关联。H.pylori感染人体后,寄居在胃黏膜上皮的黏液层,而TLR9可以识别H.pylori非甲基化的双链DNA分子[7]。研究[8]表明TLR9通过cag岛激活可以改变H.pylori感染环境中的恶性风险,而且H.pylori感染与胃黏膜中TLR9的表达增加有关[9]。因此,TLR9基因多态性可能在H.pylori感染和临床结果的转归中起到重要作用。基于此,在本研究中,课题组利用PCR-RFLP方法检测包头汉族人群中H.pylori感染与TLR9 rs187084、rs352140、rs164640的关系,发现在codominant遗传模式下,携带rs187084 TC基因型者较TT基因型者H.pylori感染风险降低;SNP rs352140与H.pylori感染无关联;在dominant遗传模式下,携带rs164640 AG+GG基因型者较AA基因型者H.pylori感染风险增加。
rs187084位于TLR9启动子区,一些研究显示了其与某些疾病的关系,如林茂虎 等[10]发现rs187084携带C等位基因与鲍氏不动杆菌感染有关联。也有研究显示其与一些疾病无关,如卫美蓉[11]和石源源 等[12]研究显示rs187084与胃癌的易感性无关。课题组研究结果显示与TT基因型相比,rs187084携带TC基因型可使H.pylori感染的风险降低0.520倍(95%CI=0.329~0.820)。然而关于rs187084与H.pylori感染的关系还未见报道,因此本研究需要扩大样本量进一步验证。
TLR9有两个外显子,第二外显子是主要编码区。rs352140位于第二外显子,本研究结果显示:rs352140与H.pylori感染没有关联,这与舒颖 等[13]研究结果一致。Loganathan et al[14]研究发现TLR9 rs352140多态性是影响印度泰米尔人H.pylori感染的疾病易感性和临床表现的潜在遗传风险因素,结果的差异可能是由于研究人群的种族不同造成。本研究还检测了TLR9基因的3′端下游的SNP rs164640与H.pylori感染的关系,结果显示,rs164640与H.pylori感染相关联。在dominant遗传模式下,与野生型AA基因型相比,携带AG+GG基因型者H.pylori感染风险增加1.569倍(95%CI=1.126~2.187)。目前,关于SNP rs164640与H.pylori感染的关系尚未见报道,所以,课题组的研究还需要进一步验证。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
当代陕西(2019年15期)2019-09-02 01:52:00
学苑创造·A版(2018年11期)2018-02-01 06:29:20
读者(2017年5期)2017-02-15 18:04:18
西南农业学报(2016年6期)2016-04-16 05:12:47
法医学杂志(2015年4期)2016-01-06 12:36:36
现代检验医学杂志(2015年6期)2015-02-06 01:44:02
实验动物与比较医学(2014年5期)2014-02-28 14:53:10
河南医学研究(2014年7期)2014-02-27 14:53:42
中国糖料(2013年1期)2013-01-22 12:28:23
