
SiLi5+团簇的结构及储氢性能理论研究
2020-01-10 03:32:06方子剑冯五强林雪麒温在国
四川大学学报(自然科学版) 2020年1期
阮 文, 方子剑, 冯五强, 林雪麒, 温在国
(井冈山大学数理学院, 吉安 343009)
1 引 言
氢是宇宙中最丰富的元素, 由于其可再生、能量密度高、易燃性强等特点,可作为一种高效的燃料物质, 且燃烧时不会产生二氧化碳和其他有害气体. 对于氢的利用可以进一步减少世界对石化燃料的依赖,有利于解决全球能源危机和环境污染问题, 被视为取代石化燃料最好的选择之一. 尽管作为一种很有前途的能源,然而氢燃料的使用必须克服储存方面的难题. 因此,人们致力于寻找和研究安全、经济的储氢材料. 传统的储氢技术是在低温液化和高压的钢瓶中储存的,这在车辆运输过程中很不安全且成本高昂,并不是理想的储氢方式. 由此,人们想到了采用固态材料来吸附氢分子以达到储氢的目的. 但要在室温和中压下获得可逆的储氢效果,氢与固态材料的相互作用能应大于物理结合能,而小于化学结合能. 如过渡金属可以经由Kubas相互作用来结合氢分子,即氢分子的反键轨道和过渡金属原子的d轨道构成反键作用,使相互作用能介于物理作用和化学作用之间. 理想化的储氢材料还应满足一些其他的条件,如较高的体积/重量密度、可回收性和质料的本钱效益低等.
迄今为止,人们对可能的储氢材料进行了大量的研究,如碳的多种同素异形体、硼纳米管、硅纳米团簇等. 它们的共同特点是比表面积大,有利于氢的吸附. 但是,单一的纳米结构与氢分子结合能力太弱,并不能有效地储氢. 实验表明, 碳纳米材料在室温下的储氢密度仅仅为0.43 wt%[1]. 为了克服单一的纳米结构储氢能力弱的问题,尝试在这些纳米材料表面采用金属原子进行掺杂(或修饰),这样有效地提高了材料的储氢能力[1-3]. 但在不断增加修饰金属原子数量后,这些金属原子会在纳米材料的表面产生聚合,这种聚合不利于材料储氢的循环利用,同时也增加了合成这些材料的困难性[4]. 由于碱金属的内聚力较小,利用碱金属原子来修饰, 可避免纳米材料表面金属的聚合, 并达到提高其储氢性能的效果[5-7]. 如Tai等人[5]发现B6Li8团簇具有很高的稳定性,其理论储氢极限可以达到24 wt%;又如平面星形Li6Si6团簇[6], 其理论储氢极限也达14.72 wt%. 在这些系统中,Li原子通过静电相互作用与氢分子相结合.
众所周知,自然界中广泛分布着Si元素,除了用于制造常规的半导体器件,最近被广泛应用于储氢材料的研究与开发.如Wang等[10]采用K原子修饰硅平面富勒烯, 理论储氢容量达6.13 wt%; 阮文等研究了Li原子修饰笼型Si5团簇的结构和储氢性能[11]、平面星形Li6Si6团簇的结构及其储氢性能[6]. 这些团簇材料的特点均以Si作为基础结构,在Si原子所构成的结构外,采用碱金属原子进行修饰,达到吸氢的目的. 最重要的一点是,这些结构的储氢能力都达到并超过了美国能源部(DOE)制定的实现氢能实用化的目标[12]:到2010年, 在室温以及安全压力下储氢量达6.5 wt%; 至2015年储氢量达9.0 wt%.
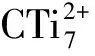
2 计算方法
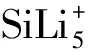
(1)
式中,E(SiLi5+)、E(SiLi5+·mH2)和E(H2)分别代表SiLi5+、SiLi5+·mH2团簇和氢分子的基态能量.

(a)
(b)
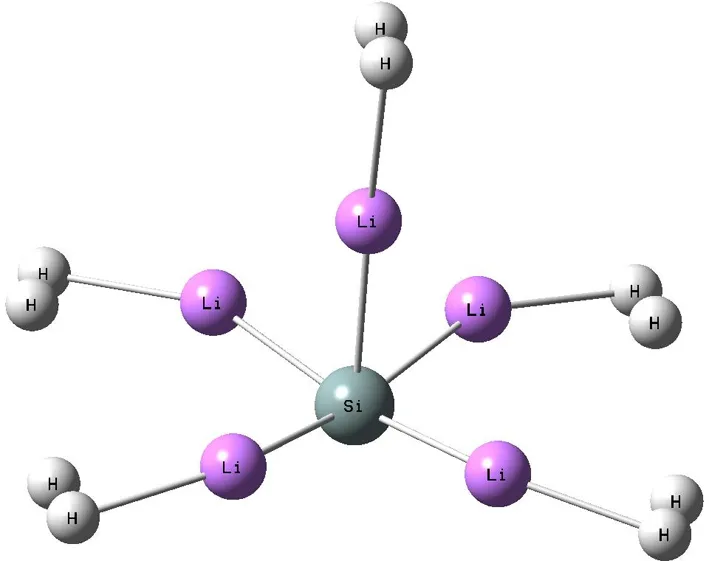
(a)
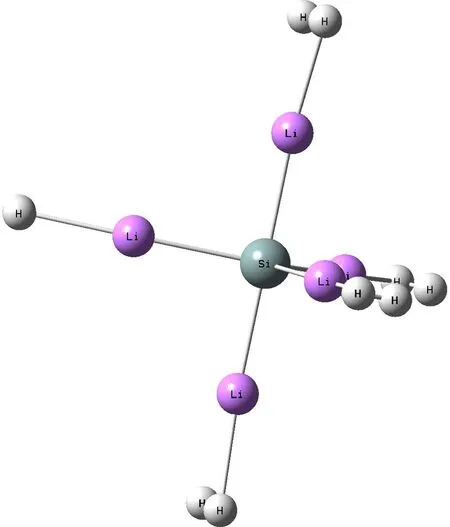
(b)
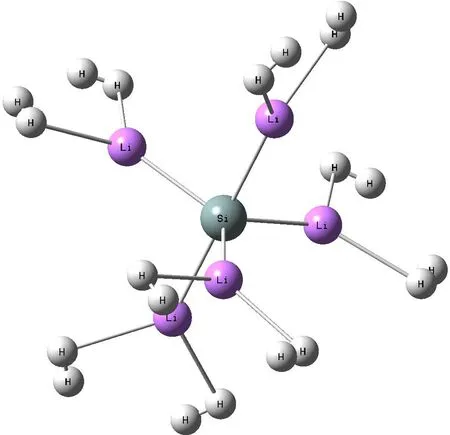
图3 SiLi5+·10H2的稳定结构

图4 SiLi5+·15H2的稳定结构

图5 SiLi5+·20H2的稳定结构
3 计算结果和分析
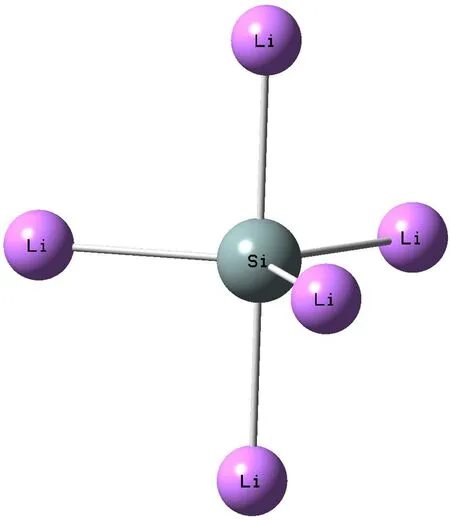
在研究SiLi5+结构的储氢性能之前,我们分别对SiLi5和SiLi5+团簇体系进行计算.考虑Li原子在Si周围的可能位置,计算了各种可能的构型,发现3个锂原子在一个平面内成120°均匀等距离分布,剩余2个锂原子分别分布在垂直于3个锂所形成的平面的两侧,并且也是等距离分布的,构成了一个三角双锥的六面体结构. 这种分布避免了锂原子之间的团簇化,由此得到了稳定的SiLi5和SiLi5+空间结构(如图1),计算结果表明SiLi5+团簇的前线轨道能隙为2.684 eV,而SiLi5团簇的前线轨道能隙为1.363 eV;且通过频率计算发现SiLi5团簇结构存在两个虚频,而SiLi5+团簇结构不存在虚频. 进一步研究当H2分子吸附时,SiLi5与H2分子相互作用后,其结构发生了严重变形,SiLi5主体部分变成了四角单锥结构,如图2(a). 很明显SiLi5三角双锥结构的动力学稳定性较差,不是一个稳定的结构,不能作为高容量的储氢材料. 比较而言,SiLi5+三角双锥结构的团簇具有高度的动力学稳定性,故接下来的工作只研究SiLi5+团簇材料的储氢性能.
为了研究SiLi5+团簇材料的储氢性能,分别考虑的从1个到4个氢分子吸附在每个锂原子周围的情况. 结果显示,每个锂原子周围最多可以吸附3个氢分子. 优化过后的SiLi5+·mH2(m=5, 10, 15, 20)结构分别如图2b、3、4和5.
由图可知,在三角双锥SiLi5+团簇材料结构中,每个锂原子能有效吸附3个氢分子(如图4). 当每个锂原子吸附到第四个氢分子时,发现每个锂原子周围都有一个氢分子被排斥出去了,其Li-H键长为4.85 Å以上(如图5),说明在三角双锥SiLi5+团簇结构中,每个锂原子最多只可有效吸附3个氢分子. 其中吸附1个氢分子时,氢分子吸附在Li原子的顶位,氢分子键与Li-Si连线垂直;吸附2个氢分子时,氢分子分布在锂原子两侧,氢分子键与Li-Si连线成一定夹角;吸附3个氢时,氢分子在空间中成对称结构分布在锂原子周围,且与Li-Si连线夹角基本相同.
分析氢分子能够在SiLi5+团簇周围吸附的原因:主要是由于锂原子和硅原子之间的相互作用,使电荷发生重新分布,导致锂原子部分的电荷转移到了硅原子上,带正电的锂原子和带负电的硅原子产生了静电场,导致了氢分子产生极化,进而通过静电相互作用而吸附在Li原子周围. 并且在氢分子吸附的过程中,氢分子有少量的电子转移到锂原子上,氢与锂原子之间也表现出了微弱的离子相互作用,这也是氢分子能够在SiLi5+团簇周围吸附的一个次要原因.
计算SiLi5+·mH2的主要结构参数列于表1,由表1可知,随着吸附氢分子数目的增加,氢分子与锂原子的平均距离逐渐增加,由此可以得出随着氢分子数目的增加,氢分子与锂原子间的相互作用呈现出逐渐减弱的趋势. 这可以从锂原子和硅原子上的电荷分布上得到解释,随着吸附氢分子数目的增加,锂原子上分布的正电荷和硅原子上分布的负电荷都在减少,表明由此产生的静电场强度在减小,导致了氢分子产生极化的强度减小,氢分子与锂原子间的相互作用必然就要减弱. 通过氢分子在SiLi5+结构中的平均吸附能计算,氢分子的平均吸附能从2.621减小至1.362 kcal·mol-1,恰好说明了随着吸附的氢分子数目的增加,锂原子对氢分子的束缚能力逐渐下降.
表1SiLi5+·mH2团簇结构参数:平均H2-Li、H-H键长,氢吸附能和电荷分布
Tab.1 The structural parameters of SiLi5+·mH2clusters: average H2-Li, H-H bond length, hydrogen adsorption energy and H2, Li and Si charge distribution

ClustersdH2-Li/ÅdH-H/ÅEad/(kcal·mol-1)Average chargeson H2/a. u.Average chargeson Li/a.u.Charges onSi/a.u.SiLi5+0.790-2.950SiLi5+ ·5H22.1050. 7492.6210.0380.670-2.542SiLi5+ ·10H22.2210. 7521.6280.0370.602-2.368SiLi5+ ·15H22.2170. 7511.3620.0520.425-1.915
在相同的计算方法和基组水平下,我们计算得出自由氢分子的键长为0.744 Å.从表1中可见,随着氢分子在SiLi5+团簇表面吸附,氢分子平均键长在0.749 ~0.752 Å范围内变化,都比自由氢分子的键长要长一些. 这是由于吸附过程中氢分子中有少量成键轨道上的电荷转移到了锂原子,削弱了氢分子成键轨道的吸引作用,由此增大了氢分子中原子间的库仑排斥作用,使得氢原子间的键长比自由状态下的氢分子的键长有所增大,但并未完全断开,形成准分子形式吸附在锂原子周围.
当继续增加吸附氢分子数量时,发现原本排列在每个锂原子周围的氢分子,最终出现一个氢分子被排斥出去的情况,其与最近的锂原子距离在4.85 Å以上(如图5).这说明当吸附氢分子数量继续增加时,SiLi5+结构所产生的静电场不能够有效束缚住更多的氢分子,所以会出现氢分子远离锂原子的情况.
4 结 论
采用密度泛函理论研究了SiLi5+团簇的结构及其与氢分子的相互作用.结果显示,5个锂原子与中间的硅原子直接成键,形成三角双锥稳定结构. 锂原子的加入,可以改善单一硅原子形成团簇的吸氢性能,氢分子在锂原子周围的吸附方式符合预期的吸附方式,即介于物理吸附和化学吸附之间,其吸附机理主要是静电极化机制. 当每个锂原子吸附3个及以下氢分子时,氢分子既不解离也不会脱附. 理论计算SiLi5+团簇的质量储氢分数为32.3 wt%,并且氢分子的平均吸附能变化范围为1.36~2.62 kcal·mol-1.较高的储氢密度以及合适的吸附能表明SiLi5+团簇结构在近室温环境下有可能成为一种潜在的储氢材料.
猜你喜欢
实验室研究与探索(2022年5期)2022-09-01 10:10:12
中国特种设备安全(2022年4期)2022-07-08 02:41:40
中国特种设备安全(2022年4期)2022-07-08 02:41:28
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
化工装备技术(2017年4期)2017-09-01 15:58:21
信息记录材料(2016年4期)2016-03-11 15:22:31
材料科学与工程学报(2016年5期)2016-02-27 07:11:37
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
断块油气田(2014年5期)2014-03-11 15:33:49
