
一种测定燃香可迁移元素含量的方法
2020-01-03 10:34刘中洁
质量技术监督研究 2019年6期
刘中洁
(国家燃香类产品质量监督检验中心(福建),福建 泉州 362000)
燃香的主要原材料是植物粉末。可迁移元素是植物的重要污染物之一,主要来源于土壤、自身主动吸收、工业“三废”、农药肥料污染等[1]。通过生物富集作用,As、Cd、Hg、Pb等在植物体内累积,其半衰期长,不易分解,达到一定数量,便会呈现毒性作用[1]。可迁移元素含量超标对人身安全、环境保护都将构成重大威胁。因此,准确测定燃香可迁移元素含量很有必要。
2011年,国家制订了GB 26386-2011《燃香类产品安全通用技术条件》[2],对燃香有害物质做了最大限值规定,同时制订了GB/T26393-2011《燃香类产品有害物质测试方法》[3](以下简称“国标法”) 。在可迁移元素含量测定方法方面,国标法假定燃香被人吞咽后与37℃的0.07mol/L盐酸模拟胃液接触,动态迁移1h,静态迁移1h,然后测定萃取的迁移量[4]。其前处理方法是稀盐酸萃取法,测定的并非可迁移元素总量,而是模拟胃液中的迁移量。这个前处理方法存在多处缺陷。
样品前处理带来的误差占分析结果误差的三分之一以上[5],选择合适的前处理方法特别重要。目前,微波消解是分析测定可迁移元素含量最为常用的前处理方法。微波消解制样容易控制,产生的酸雾少,能有效降低环境污染,减轻对人体危害。而且微波消解试验重复性好,使用密闭消解罐使常规法易挥发元素As、Cr、Hg、Pb等不损失,可获得很好的精密度与准确度。因此,文中建立了微波消解-等离子体发射光谱法(以下简称“微消法”)用以测定燃香可迁移元素含量,并对该方法的适用性进行了实验室质控分析讨论。
1 实验
1.1 试剂和材料
(1)硝酸(HNO3):优级纯。
(2)有证单元素标准物质(1000mg/L):As、Cd、Cr、Hg、Pb。
(3)标准溶液配制:吸取适量单元素标准物质,用硝酸溶液(5+95)逐级稀释配成0mg/L、0.0400mg/L、0.100mg/L、0.400mg/L、1.00mg/L、2.00mg/L混合标准系列溶液。
2 分析步骤
2.1 燃香前处理
取20g燃香(竹枝香去掉竹芯)粉碎后过80目筛,称取约0.5g过筛燃香粉末置于微波消解内罐中,加入8mL浓硝酸,摇匀,先在电热板上于120℃加热至内罐棕色烟雾消失,待冷却后盖紧罐盖置于微波消解仪中消解。消解结束后,将消解罐放在控温电热板上,于120℃加热至罐内液体约1mL,将消解液全部转移至25mL容量瓶中,用少量水冲洗罐壁,并将清洗液并入容量瓶中,用水定容至刻度,混匀,作为待测液。同时制备样品空白。
2.2 待测液的测定与结果表述
使用全谱直读电感耦合等离子体发射光谱仪依次测定混合标准系列溶液浓度,绘制校准曲线,再测定样品空白(C0)和待测液的浓度(C1),燃香可迁移元素含量X(mg/kg)计算公式[3]如下:
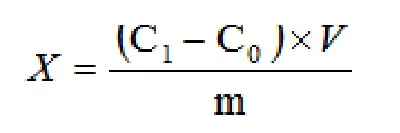
3 结果与讨论
3.1 国标法的缺陷
(1)前处理操作不严谨。国标法简单认定定容体积为100mL,未将用以调节pH的2mol/L盐酸计入定容体积内;恒温水浴振荡器的振荡频率对萃取效率有很大影响,国标法对此没有明确规定;上清液浓度不均匀,不应作为待测液。
(2)校准曲线范围过宽。国标法使用的校准浓度点最大达10.0mol/L,而GB 26386-2011《燃香类产品安全通用技术条件》规定的可迁移元素最大限量仅1.80mg/L(Pb≤90mg/kg,经公式换算后得到),校准曲线浓度范围过宽,导致测定的样品空白绝对值高,影响低浓度样品测试准确性。
(3)加标回收率偏低。经本实验室大量加标回收率试验,燃香粉末在稀盐酸溶液萃取过程中,自身会吸附一定量可迁移元素,导致加标回收率偏低,实验室质控不理想。
3.2 质控数据对比讨论
(1)关于线性及校准。按照国标法规定和微消法要求分别绘制校准曲线。两种方法线性参数见表1。由表1可见,两种方法的相关系数均大于0.997,都满足质控要求[6],二者不具明显差异性。
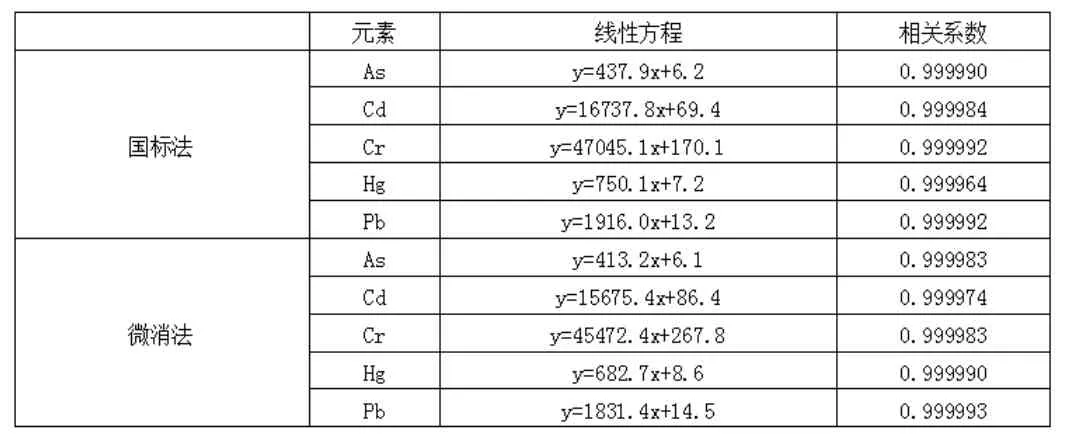
表1 线性参数
(2)关于检出限和定量限。分别对两种方法的样品空白平行测定20次,计算标准偏差[6]。两种方法检出限和定量限见表2。由表2可见,除Pb外,其他元素的检出限不具有明显差异性。微消法Pb的检出限更低,这可能是由于基体酸度较高引起的。

表2 检出限和定量限(mg/kg)
(3)含量测定对比。每个样品平行测定2次,扣除样品空白后取平均值。两种方法测定的可迁移元素含量见表3。由表3可见,两种方法测定的As、Hg含量多数处于检出限以下,这说明燃香As、Hg含量极低。两种方法测定的Cr、Pb含量均高于检出限且数值较大,可以进行统计分析:微消法测定的Cr含量是国标法的1.6倍~2.8倍,微消法测定的Pb含量是国标法的1.5倍~3.1倍。

表3 可迁移元素含量(mg/kg)
(4)关于加标回收率。两种方法加标浓度均为:As、Cd、Hg、Pb加 标 0.0400mg/L,Cr加 标0.0600mg/L。每个样品平行测定2次,取平均值。两种方法的加标回收率见表4。由表4可见,微消法的回收率基本稳定在80%~110%之间。国标法的加标回收率高低不定,As、Cd、Cr等3个元素的回收率在70%~110%之间,Hg的回收率极低,在20%以下,Pb回收率在70%以下且不具有规律性。两种方法的回收率都不固定,这可能和组成燃香的原材料有关。国标法Hg、Pb加标回收率低的原因可能是,燃香粉末在稀盐酸溶液萃取过程中吸附了加标后较高浓度的Hg、Pb,导致提取的上清液可迁移元素浓度偏低。微消法的回收率更均衡,且处于质控合理区间内。

表4 加标回收率
(5)关于精密度。实验室使用变异系数(CV)评价精密度[6],变异系数CV等价于相对标准偏差。对每个样品进行独立重复测定6次,计算相对标准偏差。两种方法的精密度见表5。由表5可见,两种方法的精密度均处于精密度期望值内[6],但微消法Hg的精密度要好于国标法。
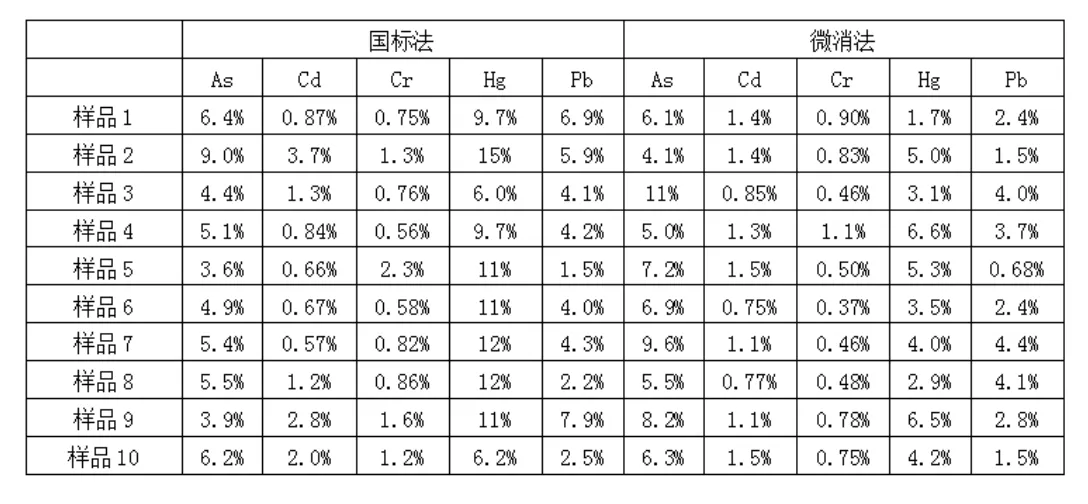
表5 精密度
3.3 微波消解前处理讨论
称取0.5g样品、使用25mL容量瓶的原因是基于公式换算关系,与国标法称取2g样品、定容到100mL相对应,深层次原因是受限于全谱直读电感耦合等离子体发射光谱仪灵敏度,使用相同的换算比例有利于测试数据对比分析。微波消解赶酸的目的是酸度高会提高待测液粘度,影响精确测定,同时也损害仪器零部件[7]。使用120℃预消解和消解后赶酸,一是因为120℃下As、Hg不会挥发[7],二是120℃下能够保证赶酸效率。
4 结论
国标法有关可迁移元素含量测定的方法内容大量参考GB 6675-2003《国家玩具安全技术规范》[8],未考虑燃香和玩具理化性质重大差别,Hg、Pb元素的加标回收率低,不同燃香的加标回收率差值大,达不到实验室质控要求,而且前处理方法存在明显缺陷,该方法不适合用于测定燃香可迁移元素含量。
国标法测定的是模拟胃液萃取的可迁移元素含量,微消法测定的则是燃香含有的可迁移元素总量,后者测定的可迁移元素含量大约是前者的1.5倍~3倍,前者仅能评价燃香对人体安全的影响,后者则兼顾燃香质量及环境评价,可全面评估燃香质量安全风险。微消法前处理步骤简单,可以做到精确定量,加标回收率更稳定且处于质控合理区间,满足实验室质控要求。综上,微消法更适合作为测定燃香可迁移元素含量的方法。
猜你喜欢
系统医学(2022年6期)2022-06-13
华人时刊(2020年21期)2021-01-14
科技视界(2018年29期)2018-12-28
中国自行车(2018年3期)2018-04-18
科学家(2016年17期)2017-10-17
中学生数理化·高三版(2017年1期)2017-04-20
汽车与安全(2016年5期)2016-12-01
科技视界(2015年15期)2015-01-16
消费电子(2014年3期)2014-03-22
