
短指(趾)症及指(趾)骨发育的分子调控机制
2019-12-24 06:43:40吕赵劼王志浩卢淑娴刘沛蓉田静
遗传 2019年12期
吕赵劼,王志浩,卢淑娴,刘沛蓉,田静
短指(趾)症及指(趾)骨发育的分子调控机制
吕赵劼,王志浩,卢淑娴,刘沛蓉,田静
西北大学生命科学学院,西部资源生物与现代生物技术教育部重点实验室,西安 710069
短指(趾)症(brachydactyly, BD)是一类指(趾)骨或掌(跖)骨的异常缩短或缺失而造成的手/足畸形病变。从临床表型上短指(趾)症可以分为单纯型短指(趾)症以及包含短指(趾)症状的综合征,其中单纯型短指(趾)症又分为5种类型:BDA、BDB、BDC、BDD和BDE,而每一类型又分为不同的亚型。作为一类重要的分子疾病家族,随着对每种短指(趾)症的深入研究,大多数单纯型短指(趾)症和部分综合征的致病基因及其分子机制逐渐被发现。虽然短指(趾)症在表型上高度多样化,但在分子水平上这些致病基因主要影响Hedgehog、NOTCH、WNT和BMP等信号传导通路。这些信号传导通路组成了一个复杂的信号调控网络,在指(趾)骨及关节的不同发育阶段发挥着不同的作用,其中BMP信号传导通路扮演着至为关键的角色。本文在目前对短指(趾)症的分类基础上,详细综述了短指(趾)症相关致病基因及所影响的信号通路等方面的最新进展,旨在探讨指(趾)骨形成的分子机制,以期为短指(趾)症的临床诊断以及人类骨骼发育的分子调控机制研究提供参考。
短指(趾)症;信号传导通路;Hedgehog;NOTCH;WNT;BMP
人的手指(脚趾)是人类进化过程中最复杂的骨骼结构之一。手指的灵活操作使人类具有处理更精细物品的能力,脚趾的灵活则给人类带来了更多的运动姿势。拥有精细复杂结构的指(趾)骨发育和功能,如同人体其他组织结构一样,受一系列复杂的分子机制调控,并对不同的病理变化高度敏感。指(趾)骨疾病在日常生活有多种呈现形式,如短(趾)指、并(趾)指、多(趾)指、软骨发育异常等,其中短指(趾)症及其综合征家族对研究指(趾)骨的正常发育有着极其重要的意义。
短指(趾)症(brachydactyly, BD)是一类指(趾)骨或掌(跖)骨的异常缩短、缺失或融合而造成的畸形病变。近年研究表明关于短指(趾)症的致病基因已达10余种并涉及到Hedgehog信号通路(hedgehog signaling pathway)、NOTCH信号通路(NOTCH signaling pathway)、WNT信号通路(wingless-related MMTV integration site signaling pathway)、BMP通路(bone morphogenetic protein signaling pathway)。本文结合Bell分类[1]和人类孟德尔遗传目录[2]对短指(趾)症的分类以及所涉及的分子信号通路进行讨论,以期为短指(趾)症的临床诊断以及人类指(趾)骨发育的分子调控机制研究提供参考。
1 短指(趾)症的分类及分子遗传学基础
随着短指(趾)症的不断研究,短指(趾)症的分类也越来越详细[3]。指(趾)骨畸形作为短指(趾)症的典型表征可在人群中表现为单纯型,也可成为复杂综合征表征的一部分[3,4]。
1.1 单纯型短指(趾)症
根据Bell分类并结合不同指(趾)骨的受累情况,单纯型短指(趾)症目前可分为A、B、C、D、E 5种分型[1,2]。BDA表现为中节指(趾)骨缩短,其中BDA1所有中节指(趾)骨均缩短或退化,偶尔与远节指(趾)骨融合,拇指和大脚趾的近节(趾)骨均有缩短;BDA2表现为第2指(趾)的中节指(趾)骨发育不全,表现出缩短或退化;BDA3的表现为第5指的中节指(趾)骨缩短,其余指(趾)骨均正常;BDA4表现为第2和第5指(趾)中节指(趾)骨短,且由于中节指(趾)骨形状异常导致远节指(趾)骨桡侧偏离,下肢受累的患者通常为第4趾中节趾骨缺失[1]。BDB具有两种分型:BDB1和BDB2。BDB1主要表现为远节和(或)中节指(趾)骨缩短或发育不全,并伴有指甲缺失;BDB2表现远节指(趾)骨发育不全,伴有远端交感神经管炎、腕/跗骨融合和局部皮肤性并指畸形[5,6]。BDC主要表现为第2、3、5指(趾)中节指(趾)骨缩短并伴有第2和/或3指(趾)指(趾)骨多节化,第1掌骨也出现缩短[7]。BDD型拇指(趾)远节指(趾)骨有不同程度的缩短,并且宽于正常表型[8]。BDE具体可分为BDE1和BDE2:BDE1限于第4掌(跖)骨的缩短;BDE2为多种掌(跖)骨缩短畸形,并且第1、3指(趾)远节指(趾)骨和第 2、5指(趾)中节指(趾)骨缩短[8,9]。
目前,已发现的单纯型短指(趾)症大多为常染色体显性遗传模式。除BDA3和BDD相对常见之外,其余单纯型短指(趾)症均极为罕见。短指(趾)症的致病基因在肢端发育过程中具有极其重要的作用,目前的研究已对部分致病基因及其分子机制有一定了解(表1)。现已发现BDA1 (MIM:112 500)致病基因为(India Hedgehog)[10]、(Growth differentiation factor-5)[11]以及(Bone Morphogenetic Protein Receptor 1B)[12]。BDA2 (MIM:112 600) 致病基因有[13][14]、(Bone Morphogenetic Protein 2)[15]等。BDB1 (MIM:113000)和BDB2 (MIM:611 377)的致病基因分别为(Receptor tyrosine kinase-like orphan receptor 2)和(Noggin)[5,6]。BDC (MIM:113 100)、BDD (MIM:113 200)的致病基因分别为[7]、(the homeobox D13)[8]。BDE1和BDE2(MIM:113 300)的致病基因分别为[8]和(parathyroid hormone like hormone)[9](表1)。
1.2 复杂短指(趾)症综合征
部分参与肢端形成的基因发生突变所引起的短指(趾)表型,可能也是某种综合征的表型之一。具有肢端缩短症状的综合征有很多,如Feingold综合征(Feingold syndrome, MIM:164 280)、Du Pan综合征(Du Pan syndrome,MIM:228900)、Robinow综合征(Robinow syndrome, MIM:268 310)、Temtamy轴前短指(趾)综合征(Temtamy preaxial brachydactyly syndrome, TPBS, MIM:605282)等(表1)。
Robinow综合征主要表现为短指与远节指(趾)骨短缩,指甲发育不良,第五指(趾)骨弯曲。Robinow综合征有两种遗传模式,常染色体显性遗传和常染色体隐性遗传,的无义突变和移码突变可以导致该疾病[16,17]。TPBS是近年来新发现的一类短指(趾)症综合征,临床诊断多表现为先天性的骨骼发育异常,1、2、3指(趾)近节指(趾)骨部分增生,常伴有智力发育迟缓、身材矮小、小头畸形、大眼、发育不良、牙齿异常、颌骨小和骨质疏松症。2010年,本课题组通过对近亲婚配的约旦家系中的患者进行DNA序列分析,发现硫酸软骨素合成酶(chondroitin sulfate synthase1, CHSY1)基因的隐性突变是造成其先天性指(趾)骨发育异常的遗传基础,并将此疾病命名为Temtamy轴前短指(趾)综合征[18]。随着TPBS致病基因的发现,越来越多的TPBS综合征在不同地区被发现,目前在埃及、土耳其、斯里兰卡、巴基斯坦和约旦等地均有该病例的报道[19]。
2 短指(趾)症相关信号通路
2.1 Hedgehog信号通路
Hedgehog信号通路由Hedgehog配体(HH)、膜蛋白受体复合物、核内转录因子和下游靶基因等4个部分组成。在高等脊椎动物中,Hedgehog配体有3种同源蛋白IHH (indiana hedgehog)、SHH (sonic hedgehog),DHH (desert hedgehog)。IHH主要表达在肥大软骨细胞之类的软骨细胞中,可以调控软骨细胞的增殖和分化,是短指(趾)症的主要致病基因之一。
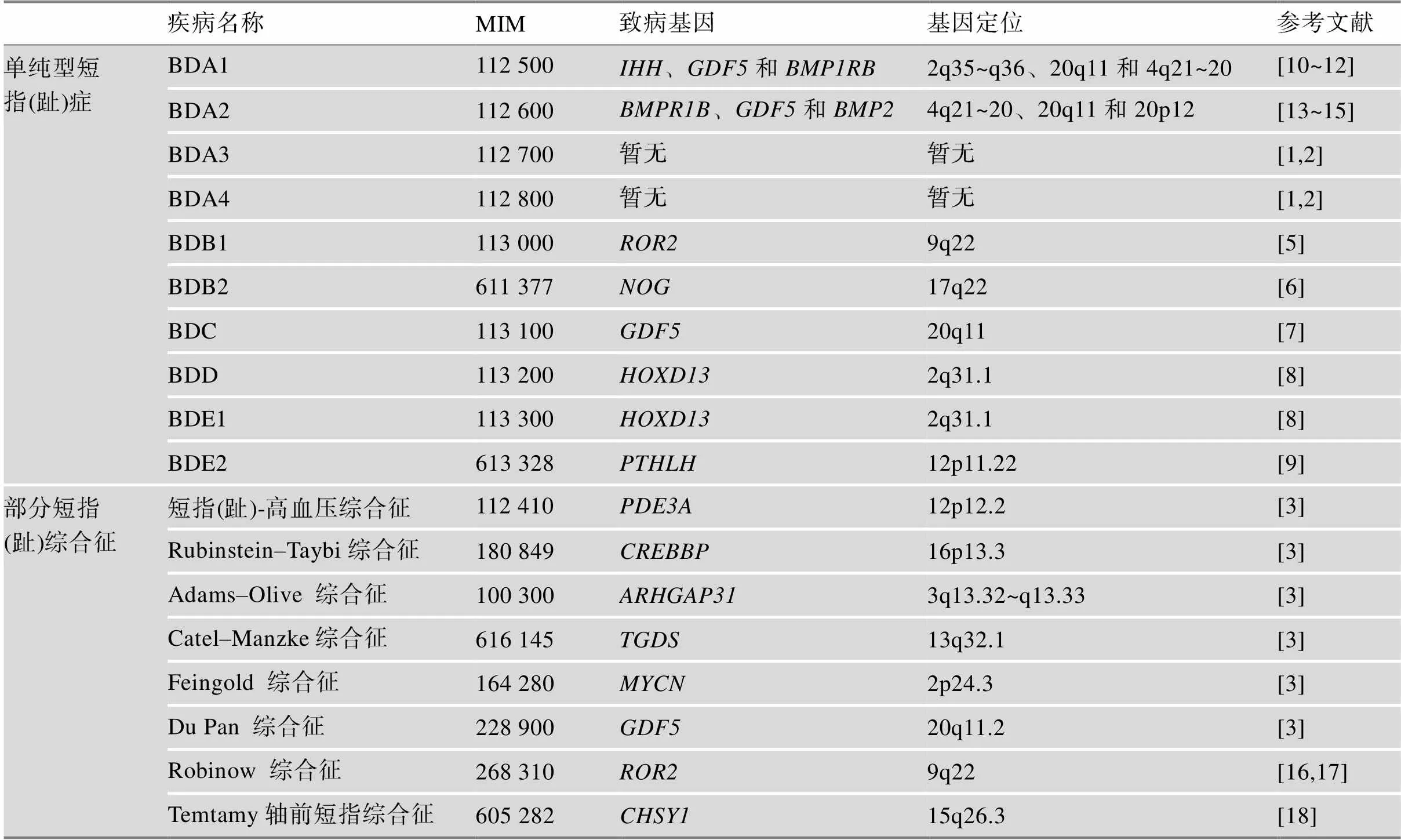
表1 单纯型短指(趾)症和部分短指(趾)症综合征
在Hedgehog信号传导通路中,当IHH配体与受体PTC结合,解除PTC对SMO的抑制作用,SMO被激活,使转录因子GLI1磷酸化,从而启动下游靶基因表达。IHH蛋白在关节周围的软骨膜中可诱导PTHLH(parathyroid hormone like hormone,甲状旁腺激素样激素)表达,PTHLH可抑制IHH的表达,与IHH形成负反馈环,从而调节Hedgehog信号通路。
IHH作为Hedgehog信号通路的配体,其突变可能会导致BDA1发生。St-Jacques等[20]通过同源重组技术敲除第一个外显子,从而得到了Ihh无义突变小鼠,并观察到小鼠肢端缩短并且出现各种成骨发育不良。上海交通大学贺林院士团队首先将BDA1的致病基因定位于染色体2q35~q36,并发现了3个错义突变,分别为E95K、D100E和E131K[5]。随后该团队Gao等[21]发现在突变体小鼠E95K/E95K中,Ihh与受体Ptch1和拮抗蛋白Hip1的结合减弱,下游转录因子Gli1激活减弱,从而下调Hedgehog信号通路。由于指(趾)骨远端祖细胞的募集减少,指(趾)骨的生长受损致使指(趾)骨的缩短。在信号通路水平上,IHH突变(E95K)导致IHH对PTC1和HIP1的结合降低,从而增加了IHH的信号范围,但导致生长板软骨细胞中短程IHH信号降低,但导致关节周围软骨膜中的远程IHH信号升高,导致PTHLH表达增加。增加的PTHLH又阻止了远端新形成的IHH表达,最终导致远端IHH急剧下降。这一系列的信号传导及蛋白表达导致了中间指(趾)骨的缩短或缺失,即产生了BDA1的表型[21]。在对IHH的3个突变(E95K、D100E和E131K)的进一步研究发现IHH的3个突变均可使Ptc1和Gli1的激活削弱。在最适诱导浓度下,突变型IHH对Gli蛋白的诱导能力大幅减弱[22]。2011年,Ma等[23]验证了3个IHH错义突变(E95K、D100E和E131K)对IHH蛋白结构、功能和结合能力的影响,全面阐述了IHH的功能与BDA1突变之间的关系[22]。E95K改变了将其位点的表面电荷由负变为正,而不改变临近结构的表面电荷。E131K可能出现两种构象:一是与E95构象一致;二是E95与K131之间形成盐桥。这种表面电荷额度改变可能会影响IHH与PTC1的相互作用。D100E突变的表面电荷分布则没有显著改变,但在D100E结构中,氨基酸残基93~98之间的区域比正常结构具有更高的灵活性。这3个突变并没有对IHH整体结构造成显著破坏,但是对钙离子的结合区域出现了细微的变化。E95K、D100E和E131K这3种突变均通过溶酶体途径导致在胞内出现不同程度的降解。IHH蛋白的细胞内稳定性可能与钙结合槽内的细微变化有关,当钙离子增多时,突变型IHH (E95K、E131K)的蛋白正常表达水平提高。这些突变可能会影响IHH的合成或分泌,是BDA1疾病机制重要的一部分。2019年,贺林院士团队Shen等[24]通过结合野生型IHH和E95K突变体进行ChIP-chip和Microarray-based 基因表达分析,提出了BDA1的Gli1介导模型,并发现E95K突变体信号传导改变了Gli1-DNA结合模式,削弱了下游基因表达,并导致细胞增殖和迁移减弱。Runx2是一种与短指(趾)症相关的基因,并且Runx2在野生型IHH和突变型IHH之间存在差异表达。Runx2相关途径是解释E95K引发的BDA1和IHH下游信号调节变化的最相关基因之一[24]。
近年来,IHH突变引发BDA1的报道也逐渐增多。2015年,Dong等[25]在3例BDA1表型中国家系中分别发现3个IHH错义突变(E95del、D100E和D100N)。2017年,Salian等[26]发现1例手足中指(趾)骨完全缺失的BDA1儿童,并发现其携带有IHH具有错义突变(D100N)。
PTHLH是Heghehog信号通路的拮抗剂,突变会导致BDE2的发生。Klopocki等[9]在5个无关联的家系中均发现PTHLH功能缺失会导致手部及足部的掌骨、跖骨、指(趾)骨缩短,第4和第5掌骨表现最为严重,即典型BDE2临床表型。此外BDE2患者也可能表现为身材矮小、迟发性牙萌出或少牙。Thomas-Teinturier等[27]在3个无血缘关系的患者中发现2种的突变。第一种为c.47_ 101þ73del128,该突变破坏了原先5号外显子的剪接位点。第二种突变为c.101_3delAAGT,该突变导致剪接位点出现异常。这两种突变均使转录异常,并过早出现终止密码子。3位患者均具有掌骨和跖骨缩短症状。
2.2 NOTCH信号通路
哺乳动物中有4种NOTCH受体,即NOTCH1~4。NOTCH配体在哺乳动物中分为2类,Delta-like (DLL)和Jagged。DLL有3种(DLL-1,3,4),Jagged有两种(Jagged-1,2)。当细胞膜上的NOTCH配体与跨膜的NOTCH受体结合,肿瘤坏死因子α转化酶(tumornecrosis factor convetingenzyme,TACE)在胞外第一次水解,NOTCH受体N端被配体表达细胞胞吞,C端则进入细胞在γ-secretase作用下二次水解,释放NOTCH受体活化形式(NICD),NOTCH信号通路被激活。激活后,NICD转运入核并在CSL蛋白作用下与DNA结合,通过调控HES1/5等转录因子,从而调控靶基因的转录、细胞的发育及分化。
关于NOTCH通路异常引起短指(趾)症的研究不多。目前研究发现,TPBS的致病基因可以通过调控NOTCH信号通路影响软骨的发育[18]。本课题组前期研究发现在突变的人类成纤维表皮原代细胞中,JAG1蛋白的表达显著增高,而在胚胎发育过程中的JAG1蛋白的异常可能会引起对NOTCH信号通路的异常调控,故表明CHSY1缺失会导致NOTCH信号通路上调[18]。本课题组通过在斑马鱼胚胎中将敲低发现Notch信号通路上调,而且敲低斑马鱼表现出与人类TPBS相似的表型,骨骼和胸鳍的发育异常,同时也会引起视网膜的过度生长。Wilson 等[28]利用同源重组技术敲除第1个外显子,从而得到–/–小鼠。Chsy1小鼠表现出软骨发育不良、骨密度降低、远节指间关节移位致使远节指(趾)骨缩短并出现分节。Filipek-Górniok等[29]通过对不同时期的斑马鱼进行阿利新兰染色,发现在早期发育阶段硫酸软骨素(chondroitin sulfate, CS)沉积于脊索鞘,通过原位杂交发现及其他硫酸软骨素糖基转移酶基因在软骨形成部位均有表达,如咽软骨、耳囊和胸鳍。目前有研究表明可能与也有密切关系。通过荧光素酶报告系统和实时定量PCR发现,的过表达会引起上调[30]。
本课题组在对1例罕见并指(趾)症家系CLS并指(趾)症(Cenani-Lenz syndactyly syndrome, MIM: 212780)的研究中也发现,其致病基因可通过NOTCH信号通路调控肢端及骨的发育[31],进一步证明了NOTCH信号通路在指(趾)骨发育中的重要作用。
2.3 WNT信号通路
WNT信号通路在物种进化上高度保守,对早期胚胎发育具有极大影响,也参与诱导骨的增殖、分化,是调控骨代谢的重要途径之一。WNT信号通路可以分为经典WNT/β-catenin信号通路、非经典WNT/PCP信号通路以及WNT/Ca2+信号通路。
经典WNT/β-catenin信号通路的传导由WNT配体与低密度脂蛋白受体5/6 (LRP5/6)、Frizzled受体结合,Dishevelled蛋白(DvL)接收信号,通过抑制APC-AXIN-GSK3β复合体,使β-catenin磷酸化水平减少,非磷酸化的β-catenin水平增加,在细胞核内与TCF/LEF结合,调控下游靶基因的转录过程。经典WNT/β-catenin信号通路通过调节间充质干细胞向成骨细胞和破骨细胞分化而使这个过程处于动态平衡。但通路的过度激活或抑制均会打破这种平衡,导致部分骨类疾病,如硬骨病和骨质疏松等[32]。
由WNT信号通路传导异常而引起的短指(趾)症目前主要为BDB1和Robinow综合征,是由基因的缺失导致[15,16]。Huang等[33]发现了一个具有BDB1表型的三代中国家系,并发现1个新的杂合子碱基缺失(c.1396–1398delaa)。这种突变导致出现一种截短的ROR2蛋白和一种具有57个氨基酸的多肽片段。Dong等[25]在1例BDB1表型中国家系发现了基因的一种新的杂合突变(S758x)。此外,基因突变导致BDB1也在土耳其、德国、巴基斯坦、中国、葡萄牙、威尔士、约旦和沙特阿拉伯等地均有报道[33]。
基因编码酪氨酸激酶受体,在WNT信号通路中主要与WNT/Ca2+信号通路中的WNT5A结合调控经典WNT/β-catenin信号通路和WNT/Ca2+信号通路平衡与正常传导。ROR2的半胱氨酸富集区与WNT5A结合,对WNT/β-catenin信号通路起负调节作用。Oishi等[34]发现–/–小鼠身材矮小,四肢和尾部均缩短,肺部和生殖器官发育不良,并且出现室间隔缺损。Oldridge等[35]在小鼠基因中插入,导致小鼠基因功能异常,软骨无法正常生成以及中间指(趾)骨缺失。德国学者Witte等[36]在W79X/W79X小鼠突变体的BDB1模型中发现间充质细胞向软骨细胞分化受阻,导致指(趾)骨无法正常延长。在指(趾)骨形成区处,BMP/pSMAD1/5/8信号减少,并且在突变体小鼠E13.5胚胎中发现远节指(趾)骨间充质细胞中Ihh表达减少,表明基因与BMP信号通路和IHH信号通路也存在密切关系。
2.4 BMP信号通路
BMP信号通路在骨的发育中占据重要的中心位置,位于指(趾)骨发育调控信号分子网络的中枢。BMP是TGF-β (转化生长因子)超家族中最大的蛋白家族,正向调节骨的增殖、分化以及形成。BMPs蛋白是BMP信号通路中的配体,包括BMP1~15。BMP受体有两种类型,Ⅰ型BMP受体(BMPR1)和Ⅱ型BMP受体(BMPR2)。Noggin(NOG)和Chordin (CHRD)是BMP信号通路上主要的抑制剂。在BMP信号通路传到中,BMPs与位于细胞膜上的BMPR1和BMPR2结合,使BMPR1和BMPR2激活。活化的BMPR1与下游SMAD1/5/8 蛋白作用并使其磷酸化。SMAD1/5/8 蛋白磷酸化后与SMAD4结合入核并在核中积累,在ID等转录因子作用下调节靶基因表达。
BMP信号通路上的多数元件均与短指(趾)症密切相关。下游调控区突变的重复序列会引发BDA2。Dathe等[10]在德裔巴西家系和欧洲家系中检查了21名患者,发现基因下游110 kb处有高度保守的重复序列。Su 等[37]在一个中国BDA2家系中确定了基因下游110 kb处存在4.6 kb的杂合重复序列。Liu等[38]通过全基因组连锁分析在一个中国BDA2家族定位到了致病基因,在基因下游调控区发现一个4671 bp的重复序列。2018年,Wang等[39]在一个BDA2表型的中国家系中同样发现了基因下游具有4671 bp重复序列,但在该家系未患病的人和正常人群中并未发现,并且该家系所有成员的基因、和的所有外显子并无突变。这些均说明位于下游的保守重复序列对于基因功能存在远程调控作用。
GDF5蛋白是BMP信号通路中的配体,也称为BMP14。GDF5蛋白广泛表达于指(趾)间关节细胞以及关节周围的祖细胞中,参与关节形成。突变会引发BDA1、BDA2、BDB2和BDC。Byrnes等[11]在一个法国加拿大家庭中发现了3个BDA1患者,其致病基因为的纯合错义突变R399C。Degenkolbe等[40]在一个墨西哥家庭的3代人中发现Ⅱ型多发性骨性连接综合征(multiple synostoses syndrome 2, SYNS2, MIM:610017)和BDA1的患者,均为错义突变W414R,该突变位于GDF5与NOG、BMPR1的结合部位,致使信号缺失,而信号减少。
突变也可能导致BDA2。Seemann等[14]在一个具有BDA2遗传的家族中鉴定基因的错义突变L441P,他们认为BDA2的表型是由配体GDF5和受体BMPR1的结合作用受抑制引起的。Plöger等[41]在一个BDA2家族六代14名患者中发现的错义突变R380Q。他们认为R380Q突变导致蛋白剪切异常,致使GDF5功能降低。Kjaer等[42]通过对一个曾发现患BDA2的丹麦/挪威大型家族更新族谱并在37位患者中进行分析,发现错义突变I441P,并且预测该突变存在于与BMPR1的结合位点。2018年,Khan等[43]也在巴基斯坦BDA2家系中发现基因突变[43]。
突变也往往导致BDC型短指(趾)症由于突变所引发的BDC,往往会表现出分化多个关节的症状。Everman等[44]在一个BDC家族中发现12号染色体基因上插入了23 bp。Schwabe等[7]在一个大型近亲结婚的土耳其家庭中发现该家族内所有的BDC患者均为M173V杂合突变。
BMPR1和BMPR2作为BMP信号通路的受体,在软骨细胞的聚集处有较高的表达。的突变也可能导致BDA1和BDA2表型。Racacho等[12]在2名类似BDA1的无亲缘关系的儿童中未发生突变,但发现基因突变,一例为杂合错义突变K325N,另一例为杂合剪切突变。突变也会导致BDA2发生。Lehmann等[13]在两个无亲缘关系的患有BDA2的德国家庭中发现了基因的杂合突变I200K和R486W。他们通过用微团培养系统进行功能分析表明,两种突变受体都能强烈抑制软骨形成。通过逆转录病毒感染鸡胚得到过表达的鸡胚,其表现出与人类指(趾)骨缩短极其相似的短指(趾)表型或会导致整个肢体的严重发育不足。
NOG蛋白是BMP信号通路的拮抗剂,基因的突变会引起BDB2。Lehmann等[6]在德国、土耳其、丹麦、伊朗、英国及北美6个BDB2家系中发现致病基因为,并鉴定出6种不同的杂合错义突变P35A、P35S、A36P、E48K、R167G和P187S。2015年,Ishino等[45]在一个五代日本家系中发现所有BDB2患者均为的错义突变C228G,该突变破坏了NOG蛋白的FingerⅡ中半胱氨酸之间连结,致使FingerⅡ结构发生严重破坏。此外,在巴基斯坦的BDB2家系中也发现其致病基因为[43]。在BDB2中,的突变使NOG与BMP和GDF5的结合能力发生异常,从而破坏了BMP信号通路的稳定,使得指(趾)骨的软骨发育异常,表现出短指(趾)的表型。Brunet等[46]通过同源重组技术得到突变体小鼠表现为在椎骨、肋骨和四肢上均有明显的缺陷,四肢短粗,关节出现融合,表型与人类BDB2相似。
有报道认为TPBS短指(趾)综合征的致病基因也参与BMP信号通路的调控,但参与BMP信号通路的具体细节仍尚未可知[47]。
3 短指(趾)症及指(趾)骨发育中的信号调控网络
Hedgehog、NOTCH、WNT、BMP信号通路均与骨骼发育密切相关,并且构成了一个复杂又庞大的信号通路网络。BMP信号通路在指(趾)骨发育中处于中枢位置,Hedgehog、NOTCH和WNT信号通路均与BMP信号通路相互作用(图1)。
Hedgehog信号通路作为指(趾)骨延长以及调控软骨细胞分化和增殖的不可或缺的一部分,与BMP信号通路的紧密交流也成为了骨骼正常发育的基础。IHH在BMP信号通路中具有多种调节功能。当软骨膜中进行骨生成过程中,IHH信号通路可以通过GLI激活BMPs定向的骨-软骨生成作用[48]。IHH的缺失则会减少BMP的表达量。另一方面,BMP信号通路也参与到IHH信号通路的调节中。在成骨细胞分化过程中,BMP2也可诱导IHH的表达,并下调PTC1的表达[49]。BMP/Smad信号可以直接激活IHH启动子。在生长板的发育过程中,IHH信号通路与PTHLH的调节保持软骨细胞正常增殖,有利于长骨的延伸。Zhang等[50]发现,在Smad4的t突变体小鼠中,Ihh、Ptc1和Pth1r的表达量均降低。Zhang等[51]在组成性激活突变型、组成性激活突变型、以及组成性激活突变型的过表达的鸡胚的肢芽中发现和的表达量上升。Minina等[52]在培养小鼠肢体的外植体中发现,Bmp2可以诱导的表达,而Noggin则会抑制的表达。

图1 短指(趾)症致病基因参与信号通路调控网络
短指(趾)症致病基因参与的信号通路主要为Hedgehog、WNT、NOTCH、BMP4条信号通路。信号通路相互调控的解释见正文。大部分短指(趾)症致病基因存在于BMP信号通路中,而不直接参与BMP信号通路的短指(趾)症致病基因可通过分子网络调控将4条信号通路联系起来,最终共同影响指(趾)骨发育进程。文中对信号通路具有抑制作用的部分用红色线条表示;对信号通路具有促进作用的用绿色箭头表示。
WNT信号通路除参与骨骼发育外,也参与到骨细胞的分化、增殖、凋亡以及维持骨量稳定等多个生理过程。WNT信号通路与BMP信号通路在指(趾)骨发育也有着紧密的联系。BMPs蛋白对WNT信号通路的调节具有双重作用。BMP蛋白可以通过MAPK和SMAD信号通路传导,从而抑制WNT信号通路传导,调节骨的正常生理发育,并在成骨细胞中对骨量进行负调节[53,54]。在WNT信号通路中Dvl起到接收受体传导的信号,并将其信号传导至下游引起β-catenin磷酸化水平降低。而BMP2则可诱导Smad1-Dvl1复合物形成,限制了WNT信号通路的传导[55]。当ROR2与WNT5A结合后调节WNT/β-catenin信号通路,同时在软骨形成中拮抗BMP信号传导。此外,ROR2通过抑制Smad1/5/9信号传导和激活Smad非依赖性途径来调节GDF5信号传导。因此当ROR2功能异常时,往往致使指(趾)骨发育异常,从而导致短指(趾)症BDB2或Rinbown综合征的发生。BMPs同时也可以与WNT信号协同作用,共同促进成骨细胞和软骨细胞的分化,保证骨骼,尤其是指(趾)骨的正常发育。BMP2可以通过诱导WNT蛋白的表达促进经典WNT信号通路[56]。Smad和Tcf/Lef/β-catenin复合体在核内启动子上相互作用,使基因、和表达增高,促进成骨细胞分化[57]。
NOTCH信号通路与BMP信号通路也存在着密切的联系,但NOTCH信号通路与BMP的在骨骼发育,尤其是指(趾)骨发育中的关联研究不多。2010年,本课题组在TPBS中发现,CHSY1可以通过抑制NOTCH信号通路影响指(趾)骨的发育[18]。Li等[47]在TPBS中则发现CHSY1可以通过抑制BMP信号通路影响指(趾)骨发育。因此,CHSY1很可能在BMP信号通路和NOTCH信号通路之间,起到重要的沟通作用。
4 结语与展望
在对短指(趾)症百年的研究过程中,短指(趾)症的分类、发病机制大多已被清楚认识,并建立起了短指(趾)症相关的研究体系。在短指(趾)症的治疗方面,尚无针对所有短指(趾)症类型的标准术式[58]。在能改善患者手脚功能及外形的前提下仅可以通过整形外科手术进行治疗,但在通常情况下,该类手术并非绝对必要[58]。目前对于单纯型短指(趾)症的分类,主要为Bell分类。由于Bell分类主要是以解剖学为基础,但部分短指(趾)症的分类与分子遗传学可能不完全相符,因此,在Bell分类的基础上结合指(趾)骨发育的分子遗传背景可能进一步优化短指症的诊断和分类。短指(趾)症是由于指(趾)骨的发育异常引起的缩短畸形,因此短指(趾)症在研究指(趾)骨发育方面是一个极好的模型。短指(趾)症作为由基因突变引起的先天性遗传病,各类短指(趾)症的致病基因以BMP信号通路为核心,并通过Hedgehog、NOTCH、WNT等信号通路对指(趾)骨发育起到决定性影响。随着短指(趾)症在分子水平上的研究,指(趾)骨发育的过程有望得到近一步细致的探索,并且可以通过更多分子水平上的手段得到更多对于短指(趾)症治疗的新思路和新方法。
[1] Temtamy SA, Aglan MS. Brachydactyly., 2008, 3: 15.
[2] OMIM. Online Mendelian Inheritance in Man: http:// www.omim.org/.
[3] Bonafe L, Cormier-Daire V, Hall C, Lachman R, Mortier G, Mundlos S, Nishimura G, Sangiorgi L, Savarirayan R, Sillence D, Spranger J, Superti-Furga A, Warman M, Unger S. Nosology and vlassification of genetic skeletal disorders: 2015 revision., 2015, 167(12): 2869–2892.
[4] Guo Y, Liang H, Deng H. Advances in the molecular genetics of brachydactyly., 2012, 34(12): 1522–1528.虢毅, 梁卉, 邓昊. 短指/趾的分子遗传学研究进展. 遗传, 2012, 34(12): 1522–1528.
[5] Stricker S, Verhey van Wijk N, Witte F, Brieske N, Seidel K, Mundlos S. Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in brachydactyly type B and Robinow syndrome., 2006, 235(12): 3456–3465.
[6] Lehmann K, Seemann P, Silan F, Goecke TO, Irgang S, Kjaer KW, Kjaergaard S, Mahoney MJ, Morlot S, Reissner C, Kerr B, Wilkie AO, Mundlos S. A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN., 2007, 81(2): 388–396.
[7] Schwabe GC, Türkmen S, Leschik G, Palanduz S, Stöver B, Goecke TO, Mundlos S. Brachydactyly type C caused by a homozygous missense mutation in the prodomain of CDMP1., 2004, 124A(4): 356–363.
[8] Johnson D, Kan SH, Oldridge M, Trembath RC, Roche P, Esnouf RM, Giele H, Wilkie AO. Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E., 2003, 72(4): 984–997.
[9] Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, Blom E, Gillerot Y, Weigel JF, Krüger G, Hiort O, Seemann P, Mundlos S. Deletion and point mutations of PTHLH cause brachydactyly type E., 2010, 86(3): 434–439.
[10] Gao B, Guo J, She C, Shu A, Yang M, Tan Z, Yang X, Guo S, Feng G, He L. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1.2001, 28: 386–388.
[11] Byrnes AM, Racacho L, Nikkel SM, Xiao F, MacDonald H, Underhill TM, Bulman DE. Mutations in GDF5 presenting as semidominant brachydactyly A1., 2010, 31(10): 1155–1162.
[12] Racacho L, Byrnes AM, MacDonald H, Dranse HJ, Nikkel SM, Allanson J, Rosser E, Underhill TM, Bulman DE. Two novel disease-causing variants in BMPR1B are associated with brachydactyly type A1., 2015, 23(12): 1640–1645.
[13] Lehmann K, Seemann P, Stricker S, Sammar M, Meyer B, Süring K, Majewski F, Tinschert S, Grzeschik KH, Müller D, Knaus P, Nürnberg P, Mundlos S. Mutations in bone morphogenetic protein receptor 1B cause brachydactyly type A2., 2003, 100(21): 12277– 12282.
[14] Seemann P, Schwappacher R, Kjaer KW, Krakow D, Lehmann K, Dawson K, Stricker S, Pohl J, Plöger F, Staub E, Nickel J, Sebald W, Knaus P, Mundlos S. Activating and deactivating mutations in the receptor interaction site of GDF5 cause symphalangism or brachydactyly type A2., 2005, 115(9): 2373–2381.
[15] Dathe K, Kjaer KW, Brehm A, Meinecke P, Nürnberg P, Neto JC, Brunoni D, Tommerup N, Ott CE, Klopocki E, Seemann P, Mundlos S. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2., 2009, 84(4): 483–492.
[16] Afzal AR, Rajab A, Fenske CD, Oldridge M, Elanko N, Ternes-Pereira E, Tüysüz B, Murday VA, Patton MA, Wilkie AO, Jeffery S. Recessive Robinow syndrome, allelic to dominant brachydactyly type B, is caused by mutation of ROR2., 2000, 25(4): 419–422.
[17] van Bokhoven H, Celli J, Kayserili H, van Beusekom E, Balci S, Brussel W, Skovby F, Kerr B, Percin EF, Akarsu N, Brunner HG. Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome., 2000, 25(4): 423–426.
[18] Tian J, Ling L, Shboul M, Lee H, O'Connor B, Merriman B, Nelson SF, Cool S, Ababneh OH, Al-Hadidy A, Masri A, Hamamy H, Reversade B. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling., 2010, 87(6): 768–778.
[19] Temtamy S, Aglan M, Topaloglu A, Wollnik B, Amr, K, El-Badry T, Hosny G, Eldin N, Shboul M, Herdem M, Ong JX, Reversade B, Tian J. Definition of the phenotypic spectrum of Temtamy preaxial brachydactyly syndrome associated with autosomal recessive CHYS1 mutations., 2012, 1(2): 64–70.
[20] St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation., 1999, 13(16): 2072–2086.
[21] Gao B, Hu J, Stricker S, Cheung M, Ma G, Law KF, Witte F, Briscoe J, Mundlos S, He L, Cheah KS, Chan D. A mutation in Ihh that causes digit abnormalities alters its signalling capacity and range., 2009, 458(7242): 1196–1200.
[22] Guo S, Zhou J, Gao B, Hu J, Wang H, Meng J, Zhao X, Ma G, Lin C, Xiao Y, Tang W, Zhu X, Cheah KS, Feng G, Chan D, He L. Missense mutations in IHH impair Indian Hedgehog signaling in C3H10T1/2 cells: implications for brachydactyly type A1, and new targets for Hedgehog signaling., 2010, 15(1): 153–176.
[23] Ma G, Yu J, Xiao Y, Chan D, Gao B, Hu J, He Y, Guo S, Zhou J, Zhang L, Gao L, Zhang W, Kang Y, Cheah KS, Feng G, Guo X, Wang Y, Zhou CZ, He L. Indian hedgehog mutations causing brachydactyly type A1 impair Hedgehog signal transduction at multiple levels., 2011, 21(9): 1343–1357.
[24] Shen L, Ma G, Shi Y, Ruan YF, Yang XH, Wu X, Xiong YY, Wan CL, Yang C, Cai L, Xiong LK, Gong XL, He L, Qin SY. p.E95K mutation in Indian hedgehog causing brachydactyly type A1 impairs IHH/Gli1 downstream transcriptional regulation., 2019, 20(1): 10.
[25] Dong S, Wang Y, Tao S, Zheng F. Mutation screening in candidate genes in four Chinese brachydactyly families., 2015, 45(1): 94–99.
[26] Salian S, Shukla A, Nishimura G, Girisha KM. Severe form of brachydactyly Type A1 in a child with a c.298G > A mutation in IHH gene., 2017, 6(3): 177–180.
[27] Thomas-Teinturier C, Pereda A, Garin I, Diez-Lopez I, Linglart A, Silve C, de Nanclares GP. Report of two novel mutations in PTHLH associated with brachydactyly type E and literature review., 2016, 170(3): 734–742.
[28] Wilson DG, Phamluong K, Lin WY, Barck K, Carano RA, Diehl L, Peterson AS, Martin F, Solloway MJ. Chondroitin sulfate synthase 1 (Chsy1) is required for bone development and digit patterning., 2012, 363(2): 413– 425.
[29] Filipek-Górniok B, Holmborn K, Haitina T, Habicher J, Oliveira MB, Hellgren C, Eriksson I, Kjellén L, Kreuger J, Ledin J. Expression of chondroitin/dermatan sulfate glycosyltransferases during early zebrafish development., 2013, 42(8): 964–975.
[30] Guo M, Liu Z, Willen J, Shaw CP, Richard D, Jagoda E, Doxey AC, Hirschhorn J, Capellini TD. Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height., 2017, 6: e29329.
[31] Tian J, Shao JH, Liu C, Hou HY, Chou CW, Shboul M, Li GQ, El-Khateeb M, Samarah OQ, Kou Y, Chen YH, Chen MJ, Lyu Z, Chen WL, Chen YF, Sun YH, Liu YW. Deficiency of lrp4 in zebrafish and human LRP4 mutation induce aberrant activation of Jagged-Notch signaling in fin and limb development., 2019, 76(1): 163–178.
[32] Shao JH, Wang ZH, Liu C, Lyu ZJ, Tian J. LRP4 and human rare genetic diseases., 2018, 30(8): 855–861.邵金辉, 王志浩, 刘聪, 吕赵劼, 田静. LRP4与人类罕见遗传病. 生命科学, 2018, 30(8): 855–861.
[33] Huang D, Jiang S, Zhang Y, Liu X, Zhang J, He R. A new mutation in the gene ROR2 causes brachydactyly type B1., 2014, 547(1): 106–110.
[34] Oishi I, Suzuki H, Onishi N, Takada R, Kani S, Ohkawara B, Koshida I, Suzuki K, Yamada G, Schwabe GC, Mundlos S, Shibuya H, Takada S, Minami Y. The receptor tyrosine kinase Ror2 is involved in non-canonical Wnt5a/ JNK signalling pathway., 2003, 8(7): 645– 654.
[35] Oldridge M, Fortuna AM, Maringa M, Propping P, Mansour S, Pollitt C, DeChiara TM, Kimble RB, Valenzuela DM, Yancopoulos GD, Wilkie AO. Dominant mutations in ROR2, encoding an orphan receptor tyrosine kinase, cause brachydactyly type B., 2000, 24(3): 275–278.
[36] Witte F, Chan D, Economides AN, Mundlos S, Stricker S. Receptor tyrosine kinase-like orphan receptor 2 (ROR2) and Indian hedgehog regulate digit outgrowth mediated by the phalanx-forming region., 2010, 107(32): 14211–14216.
[37] Su P, Ding H, Huang D, Zhou Y, Huang W, Zhong L, Vyse TJ, Wang Y. A 4.6 kb genomic duplication on 20p12.2- 12.3 is associated with brachydactyly type A2 in a Chinese family., 2011, 48(5): 312–316.
[38] Liu XD, Gao LH, Zhao AM, Zhang R, Ji BH, Wang L, Zheng YL, Zeng BF, Valenzuela RK, He L, Ma J. Identification of duplication downstream of BMP2 in a Chinese family with Brachydactyly Type A2 (BDA2)., 2014, 9(4): e94201.
[39] Wang WB, Jia YC, Zhang Z, Xu J, Zuo RT, Kang QL. A novel duplication downstream of BMP2 in a Chinese family with Brachydactyly type A2 (BDA2)., 2018, 642: 110–115.
[40] Degenkolbe E, König J, Zimmer J, Walther M, Reißner C, Nickel J, Plöger F, Raspopovic J, Sharpe J, Dathe K, Hecht JT, Mundlos S, Doelken SC, Seemann P. A GDF5 point mutation strikes twice-causing BDA1 and SYNS2., 2013, 9(10): e1003846.
[41] Plöger F, Seemann P, Schmidt-von Kegler M, Lehmann K, Seidel J, Kjaer KW, Pohl J, Mundlos S. Brachydactyly type A2 associated with a defect in proGDF5 processing., 2008, 17(9): 1222–1233.
[42] Kjaer KW, Eiberg H, Hansen L, van der Hagen CB, Rosendahl K, Tommerup N, Mundlos S. A mutation in the receptor binding site of GDF5 causes Mohr-Wriedt brachydactyly type A2.2006, 43(3): 225–231.
[43] Khan S, Mudassir M, Khan N, Marwat A. Brachdactyly instigated as a result of mutation in GDF5 and NOG genes in pakistani population., 2018, 34(1): 82–87.
[44] Everman DB, Bartels CF, Yang Y, Yanamandra N, Goodman FR, Mendoza-Londono JR, Savarirayan R, White SM, Graham JM Jr, Gale RP, Svarch E, Newman WG, Kleckers AR, Francomano CA, Govindaiah V, Singh L, Morrison S, Thomas JT, Warman ML. The mutational spectrum of brachydactyly type C., 2002, 112(3): 291–296.
[45] Ishino T, Takeno S, Hirakawa K. Novel NOG mutation in Japanese patients with stapes ankylosis with broad thumbs and toes., 2015, 58(9): 427–432.
[46] Brunet LJ, McMahon JA, McMahon AP, Harland RM. Noggin, cartilage morphogenesis, and joint formation in the mammalian skeleton., 1998, 280(5368): 1455– 1457.
[47] Li Y, Laue K, Temtamy S, Aglan M, Kotan LD, Yigit G, Canan H, Pawlik B, Nürnberg G, Wakeling EL, Quarrell OW, Baessmann I, Lanktree MB, Yilmaz M, Hegele RA, Amr K, May KW, Nürnberg P, Topaloglu AK, Hammerschmidt M, Wollnik B. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in Chondroitin Synthase 1, a potential target of BMP signaling., 2010, 87(6): 757–767.
[48] Hojo H, Ohba S, Taniguchi K, Shirai M, Yano F, Saito T, Ikeda T, Nakajima K, Komiyama Y, Nakagata N, Suzuki K, Mishina Y, Yamada M, Konno T, Takato T, Kawaguchi H, Kambara H, Chung UI. Hedgehog-Gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium., 2013, 288(14): 9924–9932.
[49] Long F, Chung UI, Ohba S, McMahon J, Kronenberg HM, McMahon AP. IHH signaling is directly required for the osteoblast lineage in the endochondral skeleton., 2004, 131(6): 1309–1318.
[50] Zhang J, Tan X, Li W, Wang Y, Wang J, Cheng X, Yang X. Smad4 is required for the normal organization of the cartilage growth plate., 2005, 284(2): 311–322.
[51] Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O'Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development., 2003, 18(9): 1593–1604.
[52] Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP, Vortkamp A. BMP and IHH/PTHrP signaling interact to coordinate chondrocyte proliferation and differrentiation., 2001, 128(22): 4523–4534.
[53] Kamiya N, Kobayashi T, Mochida Y, Yu PB, Yamauchi M, Kronenberg HM, Mishina Y. Wnt inhibitors Dkk1 and Sost are downstream targets of BMP signaling through the type IA receptor (BMPRIA) in osteoblasts., 2010, 25(2): 200–210.
[54] Kamiya N, Ye L, Kobayashi T, Mochida Y, Yamauchi M, Kronenberg HM, Feng JQ, Mishina Y. BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway., 2008, 135(22): 3801–3811.
[55] Liu Z, Tang Y, Qiu T, Cao X, Clemens TL. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells.2006, 281(25): 17156–17163.
[56] Rawadi G, Vayssière B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop., 2003, 18(10): 1842–1853.
[57] Rodríguez-Carballo E, Ulsamer A, Susperregui AR, Manzanares-Céspedes C, Sánchez-García E, Bartrons R, Rosa JL, Ventura F. Conserved regulatory motifs in osteogenic gene promoters integrate cooperative effects of canonical Wnt and BMP pathways., 2011, 26(4): 718–729.
[58] Yang X, Wang B. Etiology, classification and treatment of brachydactyly., 2015, 11(6): 389–395.杨茜, 王斌. 先天性短指畸形的发病机制、分类及治疗进展. 组织工程与重建外科杂志, 2015, 11(6): 389–395.
Brachydactyly and the molecular mechanisms of digit formation
Zhaojie Lyu, Zhihao Wang, Shuxian Lu, Peirong Liu, Jing Tian
Brachydactyly (BD) is a type of hand/foot malformation caused by the abnormal shortening or missing phalanges and/or metacarpals/metatarsals. BD most often occurs as an isolated trait, but can also occur as part of complex malformation syndromes. According to the patterns of affected digits, isolated BD can be divided into five groups: BDA, BDB, BDC, BDD, and BDE with individual subtypes. As an important molecular disease family, the pathogenic genes and molecular mechanisms of most isolated BD forms and some complicated syndromes are elucidated. Although BDs are highly diversified in phenotypes, at the molecular levels these pathogenic genes mainly affect several important signaling pathways: Hedgehog, NOTCH, WNT and BMP. These pathways form a complex signaling network and play different roles in different stages of the digit and joint development, in which BMP signaling pathway occupies a central position. Based on the current classification of BDs, this review summarizes the latest progress in the pathogenesis of BDs and the signaling pathways involved. The purpose of this review is to explore the molecular mechanisms of digit formation, which will provide references for the clinical diagnosis of BD, and the understanding of molecular mechanism of human bone development.
brachydactyly; signaling pathways; Hedgehog; NOTCH; WNT; BMP
2019-06-27;
2019-08-09
西部资源生物与现代生物技术教育部重点实验室开放研究基金项目(编号:ZSK2018010)资助[Supported by the Opening Foundation of Key Laboratory of Resource Biology and Biotechnology in Western China (Northwest University), Ministry of Education (No. ZSK2018010)]
吕赵劼,硕士研究生,专业方向:人类遗传与发育生物学。E-mail: ZhaojieLyu@outlook.com
田静,博士,教授,研究方向:人类遗传与发育生物学。E-mail: tianjing@nwu.edu.cn
10.16288/j.yczz.19-100
2019/10/14 18:44:00
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191014.1535.001.html
(责任编委: 谢小冬)
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
中国临床医学影像杂志(2019年5期)2019-08-27 02:48:00
现代园艺(2017年21期)2018-01-03 06:41:32
罕少疾病杂志(2016年4期)2016-03-11 16:34:41
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
浙江医学(2014年17期)2014-04-13 10:13:16
