
基于XGBoost对肺鳞癌和肺腺癌的分类预测
2019-12-23 08:21冷菲李巍
首都医科大学学报 2019年6期
冷 菲 李 巍
(国家儿童医学中心 首都医科大学附属北京儿童医院 遗传与出生缺陷防治中心 北京市儿科研究所 出生缺陷遗传学研究北京市重点实验室 儿科重大疾病研究教育部重点实验室,北京100045)
肺癌是最常见的恶性肿瘤之一,2012年全球新发病例约182万例,病死率约为159万[1]。尽管近年来已经取得了治疗方法的进步,包括微创手术方法,化学疗法和靶向治疗,但肺癌患者的5年生存率大多数情况下仅为10%~20%[2]。大多数肺癌由两种主要病理亚型构成:肺鳞状细胞癌和肺腺癌,二者在临床表现上非常相似,但是在发病机制以及治疗和预后方面都有明显不同[3]。
肺鳞癌和肺腺癌来自不同的细胞,不仅在生物模式方面,而且在分子特征方面,以及最重要的治疗策略方面均存在差异。例如,激活表皮生长因子受体的突变和间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)融合蛋白的突变通常发生在肺腺癌中,而不是肺鳞癌中,使针对这些基因的药物对肺鳞癌无效[4]。因此,利用这两种不同肺癌亚型的分子特征和机制的差异预测亚型分类,可以有针对性地对不同亚型疾病进行干预,同时,通过深入研究两种亚型的主要差异特征,将有助于更深入地理解和鉴定新的肺癌治疗分子靶向策略。
本研究应用机器学习分类器,极限梯度增强算法(extreme gradient boosting,XGBoost),通过RNA表达谱区分肺鳞癌和肺腺癌患者。选择这种方法是因为它具有显著的优点,包括:可以处理缺失值,需要数据缩放,提示梯度增强算法中的有效变异,在各种竞赛中取得突出的成绩,并已成功应用于其他研究领域。通过使用XGBoost区分肺鳞癌和肺腺癌,并且识别能够区分两者的主要RNA分子。目前,没有研究使用XGBoost根据RNA特征客观的对两种亚型进行分类。
本研究利用癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库发布的高通量数据[5],构建了包含474例样本的肺鳞癌和491例样本的肺腺癌数据库,包含每个样本的RNA表达数据。接下来,鉴定了肺鳞癌和肺腺癌的差异表达RNA。之后,基于XGBoost模型开发肺鳞癌和肺腺癌分类预测模型,并对预测中的重要特征进行分析。最后基于预测中特征重要性,分析了显著影响肺鳞癌和肺腺癌分类预测的RNA分子。
1 资料与方法
1.1 数据来源
从TCGA数据门户检索肺鳞癌样本和肺腺癌样本的RNA序列数据。 TCGA数据集(https://portal.gdc.cancer.gov/)由超过2 PB的基因组数据组成,可公开获得,这种基因组信息有助于癌症研究界改进预防,诊断和治疗癌症。本研究符合TCGA提供的出版指南。由于数据来自TCGA数据库,因此道德委员会无须进一步批准。
1.2 数据集构建
从TCGA数据门户下载肺鳞癌和肺腺癌的RNA-seq数据,数据来源于IlluminaHiSeq_RNASeq测序平台;所有数据都可以免费下载。分别去除两个数据集中的正常样品,共得到474例肺鳞癌样品和491例肺腺癌肿瘤样本。去除仅在肺鳞癌样本出现或仅在肺腺癌样本中测得的mRNA,得到最终mRNA数据集。在研究中,主要使用Python和R语言编写的程序代码来分析和处理RNA数据。
1.3 数据预处理
使用Ensembl数据库(http://www.ensembl.org/index.html,版本95)鉴定了mRNA[6],排除了未包括在数据库中的mRNA。之后,删除平均读数≤1的mRNA,以过滤掉所有未表达的mRNA。
1.4 特征处理
由于mRNA数据种类太多,作为特征维度太大,需进行特征处理,即从特征中选择出特征子集。为了便于后期对分类影响的评价,将不对前期数据进行特征变换处理。本文基于基因表达量在亚型之间的差异度对特征进行筛选。
使用EdgeR对两种疾病亚型的mRNA数据进行表达差异分析,采用M值修剪均值(trimmed mean of M-values,TMM)算法进行标准化,并获得差异表达的mRNA。所有P值使用错误发现率(false discovery rate,FDR)来校正多重测试的差异有统计学意义。对于倍数变化(log2绝对值)>2且FDR为P<0.001的表达差异被认为是显著的。使用R平台中的ggplots和热图包生成火山图。删除差异无统计学意义表达的mRNA,将差异有统计学意义表达的mRNA作为模型的特征集合。
1.5 模型算法
基于XGBoost算法建立模型。不同于传统集成决策树算法,XGBoost在损失函数里加入了正则项,在控制模型的复杂度的同时,可以防止模型过拟合。即得到目标函数为:

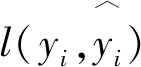
1.6 统计分析与结果评价
采用Python 3.7.1进行统计学分析。为了更加准确和全面的评估模型,采用准确率、曲线下面积(area under curve,AUC)和洛伦兹曲线(kolmogorov-smirnov,KS)作为预测结果的评价标准。
1.7 模型训练和验证
根据以上算法,建立分类模型。将处理后的数据进行多次训练,对不同参数进行多次调整,最终得到最佳结果。
2 结果
2.1 特征处理
从TCGA数据库中下载肺鳞癌和肺腺癌临床数据和mRNA表达数据,剔除正常组织样本和信息缺失样本,得到474例肺腺癌样本和491例肺鳞癌样本。分别下载474例肺腺癌肿瘤组织和491例肺鳞癌肿瘤组织中mRNA的表达量数据。基于本研究特征处理标准,得到1 099个mRNA在肺鳞癌和肺腺癌中差异有统计学意义的表达。图1通过火山图显示了-log(FDR)和logFC两个维度上所有差异表达的mRNA的分布。将所有mRNA表达水平标准化为样品平均值。剔除表达差异无统计学意义的基因,选择差异表达的基因作为模型的特征子集,建立疾病亚型预测模型。

图1 基因差异表达分析结果Fig.1 Gene differential expression analysis results
2.2 预测结果及模型性能比较
本次实验在Linux环境下进行,选择 Python 语言进行实现,将1 099个差异表达的mRNA作为特征,基于XGBoost进行建模,预测结果与数据库中真实数据进行比较,最终得出评判结果。计算分类预测的准确率为96.55%,AUC值为99.04%,Kappa值为0.92,肺鳞癌阳性预测值为0.97,肺腺癌阳性预测值为0.96(表1)。

表1 预测结果统计Tab.1 Statistics of the prediction results
为了比较模型性能,除了XGBoost模型之外,本研究同时还采用了逻辑回归算法和支持向量机算法进行建模和预测。对3种模型分别进行参数优化,利用相同方法划分训练集和测试集,分别对肺鳞癌和肺腺癌的分类进行预测,3种模型最终准确率分别为96.55%,89.97%和89.34%(表2),XGBoost模型预测准确率明显高于逻辑回归模型(χ2=37.66,P<0.001)和支持向量机模型(χ2=49.8,P<0.001),前者比后两者的准确率分别提高近6.58和7.21个百分点。 3种模型的预测AUC结果见图2。XGBoost模型的受试者操作特征(receiver operating characteristic,ROC)曲线,分别高于逻辑回归模型和支持向量机模型近4个百分点。

表2 3种预测模型预测结果比较Tab.2 Comparison of prediction results of three prediction models

图2 不同模型预测曲线下面积结果比较Fig.2 Area under curve comparison for different prediction models
为了评估模型将正、负样本区分开的程度,计算KS值(图3)XGBoost模型的KS值明显高于logistic regression模型和SVM模型,基于mRNA的肺鳞癌和肺腺癌的分类,XGBoost模型的预测性能优于另外2种模型。

图3 不同模型预测洛伦兹曲线结果比较Fig.3 Kolmogorov-Smirnov comparison of different prediction models
2.3 肺癌亚型的分子标记因子
根据特征重要性,对1 099个mRNA进行打分,权重越大代表特征对预测结果贡献越大。统计所有特征的重要性分数的分布情况(图4),发现932个mRNA特征的重要性分数为0,即大多数mRNA对疾病亚型的分类没有影响。
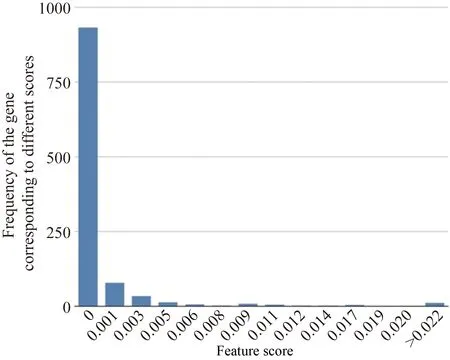
图4 基因特征重要性打分的分布情况Fig.4 Distribution of feature scores
为了进一步筛选特征,根据特征的权重对特征从大到小排序,依次累加特征,形成逐渐增大的特征子集。计算不同大小特征子集下预测结果的准确率,即这些特征对预测结果的累计贡献率,结果如图5。仅使用贡献率最大的mRNA进行预测,准确率已经达到91.5%。随着特征数目的增加,准确率逐渐升高,当特征数为11个时,准确率达到95.9%。之后,准确率的数值趋于稳定,不再根据特征数的增加而显著增长,甚至偶尔存在轻微下降的情况。

图5 不同特征子集的预测准确率Fig.5 Prediction accuracy of different feature subsets
因此,选择F-score排名前11的mRNA作为区分肺鳞癌和肺腺癌的分子标记因子,这11个mRNA的基因名称和重要性评估结果见表3。

表3 排名前11的基因名称及重要性得分Tab.3 Top 11 gene names and their importance scores
3 讨论
肺鳞癌和肺腺癌是最常见的两种非小细胞肺癌,二者临床表现相似,但致病机制和治疗预后不同,尤其存活率差异很大,而目前尚无有效的早期诊断方法。本研究基于转录组数据深入研究两种肺癌亚型的分子特征,首先通过差异分析找到1 099个在肺鳞癌和肺腺癌中表达差异有统计学意义的基因,将它们作为模型特征,之后结合XGBoost建立模型,对癌症亚型进行预测。准确率达到96%以上。基于最终建立的模型,对特征重要性进行评估。此外,还比较了XGBoost模型和其他模型的预测结果。选择了分类问题中比较经典的逻辑回归算法和支持向量机算法建立模型。
逻辑回归使用Sigmoid函数,将线性模型的结果压缩到0~1,使其拥有概率意义。支持向量机则是在特征空间中寻找使正类负类间隔最大的超平面的线性分类器。根据结果可知,这两种模型的准确率均较XGBoost偏低。由于XGBoost模型用到了损失函数的二阶泰勒展开,因此与损失函数更接近,收敛更快。并且在损失函数中加入了正则项,可以有效控制模型复杂度,防止模型过拟合。根据特征重要性评估,找到11个基因作为区分肺鳞癌和肺腺癌的分子标记因子,其中贡献前3名的基因依次是MACC1、KRT5和SPRR2E。MACC1蛋白定位于人类的7号常染色体上(7P21.1),具有广泛的生物学功能,特别是在调控恶性肿瘤的侵袭和转移等方面具有不可代替的重要功能[7]。MACC1 在多种恶性肿瘤如结肠癌、卵巢癌、肺癌、肝癌、胃癌等组织中表达异常增高,与肿瘤临床分期、有无远处转移密切相关,有作为肿瘤转移和预后判断的潜在独立指标[8]。Keratin 5,也称为KRT5,K5或CK5,是由KRT5基因在人体内编码的蛋白质。这种蛋白质涉及多种疾病,包括大疱性表皮松解症和乳腺癌和肺癌[9]。SPRR2E是染色体1q21上的人表皮分化复合物的一部分,在一项关于表皮鳞状细胞癌的研究[10]中,与正常相比,恶性角质形成细胞系中SPRR2E呈现低表达,提示终末分化缺陷,而这正是致癌转化的特征。以上研究[9-10]表明SPRR2E主要功能与皮肤角质形成细胞的分化密切相关,但与肺癌发生的作用尚不明确。有研究[11]显示支气管上皮细胞经氡染毒后,会引起SPRR2E表达量的显著变化。
综上所述,本文建立了预测肺鳞癌和肺腺癌的模型,具有较高的准确性和良好的稳定性,为其早期诊断和治疗提供理论依据。同时找到一些特征变量作为区分肺鳞癌和肺腺癌的分子标记,后续研究中将会进一步探索这些特征变量对分类的影响机制。
猜你喜欢
云南医药(2021年3期)2021-07-21
中成药(2018年8期)2018-08-29
中成药(2018年7期)2018-08-04
中华老年口腔医学杂志(2016年2期)2017-01-15
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国病理生理杂志(2015年8期)2015-12-21
中国实验动物学报(2015年3期)2015-12-16
中国当代医药(2015年30期)2015-03-01
中国当代医药(2015年17期)2015-03-01
癌变·畸变·突变(2015年3期)2015-02-27
