
Mowat-Wilson综合征5例患者临床特征分析及ZEB2基因型研究
2019-12-11 03:42姜茜,王慧,韩璐,张震,肖萍,李龙,李颀*
山西医科大学学报 2019年11期
姜 茜,王 慧,韩 璐,张 震,肖 萍,李 龙,李 颀*
(1首都儿科研究所遗传研究室,北京 100020;2首都儿科研究所附属儿童医院普通外科;3首都儿科研究所附属儿童医院病理科;*通讯作者,E-mail:l817@sina.com)
Mowat-Wilson综合征(MWS)是一种由E盒结合锌指蛋白2(ZEB2)基因突变导致的罕见常染色体显性遗传病,发病率为1/50 000-1/70 000,迄今为止全世界已报道300余例患儿[1-3]。该病临床表现多样,主要包括特殊面容合并中-重度智力发育迟缓、先天性巨结肠(HSCR)、癫痫及多种先天畸形(如生殖系统畸形、先天性心脏病、胼胝体发育不全及眼部异常等)。MWS的致病基因位于2q22.3,目前发现ZEB2基因的新发杂合破坏性突变是主要致病原因,以提前终止密码子和大片段缺失为主,亦偶见错义突变或重复突变[4,5]。MWS患儿的面容异常(如小头畸形、前额突出、眼眶深陷、眼距增宽、眉头增粗断裂、圆形鼻尖、宽鼻梁、鹰钩鼻、耳垂隆起、小下颌等)在婴幼儿时期多不明显,会随年龄增长逐渐显现,因而给早期临床诊断造成了一定困难[6,7]。近年来二代基因测序技术、比较基因组杂交技术等的发展和广泛应用为疾病分子遗传学诊断提供了重要依据。本文通过对5例MWS患儿的临床特征及基因检测结果进行分析,最终确定患儿的突变类型、分布规律及其与临床表型之间的相关性。
1 资料与方法
1.1 研究对象及资料采集
以2013-12~2019-07在首都儿科研究所附属儿童医院临床诊断为MWS的5例患儿为研究对象,其中男3例(60%),女2例(40%),4例患儿起病年龄<1岁,平均就诊年龄(12.2±19.0)月。
收集全部患儿的相关临床资料,主要包括:性别、就诊年龄、起病年龄、主要症状、家族史、体格检查结果及影像学检查结果。
1.2 分子遗传学检测
本研究通过首都儿科研究所伦理委员会的批准(伦理审查编号:SHERLL2013039),经患儿监护人签署知情同意书后取患儿及其核心家系家庭成员外周血各5 ml,提取基因组DNA。使用Nanodrop 1 000分光光度计进行质检,合格后送至北京迈基诺基因科技股份有限公司,利用Illumina NextSeq 500第二代测序仪针对172个HSCR及其综合征相关基因的外显子和侧翼序列进行高通量测序。最终5例样品的候选基因平均测序深度为238×,捕获区覆盖度达92%以上,为找出致病性点突变,参考dbSNP数据库(https://www.ncbi.nlm.nih.gov/snp)、HapMap数据库(ftp://ftp.ncbi.nlm.nih.gov/hapmap)、千人基因组数据库(1 000 genomes project,http://www.internationalgenome.org/home)、NHLBI外显子组测序项目(ESP,NHLBI exome sequencing project,http://evs.gs.washington.edu/EVS)、genome Aggregation数据库(http://gnomad-old.broadinstitute.org)和内部正常对照人群数据库,将频率小于0.001的变异视为可疑,并进一步行Sanger测序验证。应用人类基因突变数据库(http://www.hgmd.cf.ac.uk)确认已报道的致病基因位点。对于未报道过的新变异位点,应用ACMG指南标准[8]对其进行致病性评级。
2 结果
2.1 临床表现
5例(100%)患儿均存在异常面容,共有的突出表现为小下颌、眼距增宽、鸟喙样鼻小柱突出及耳垂隆起(见图1)。全部患儿均以先天性巨结肠为首诊主诉。除病例1于4月龄死于重症心衰和肺炎无法评估外,另外4例(80%)均存在智力障碍/发育迟缓。4例(80%)患儿伴先天性心脏病,2例(40%)伴生殖器官畸形,2例(40%)伴癫痫发作(见表1)。

全部5例患儿均有明显的小下颌、眼距增宽、鸟喙样鼻小柱突出及耳垂隆起
表1 5例Mowat-Wilson综合征患儿的主要临床特征及与既往文献报道病例的比较
Table 1 Major clinical features of 5 children with Mowat-Wilson syndrome and previously reported cases
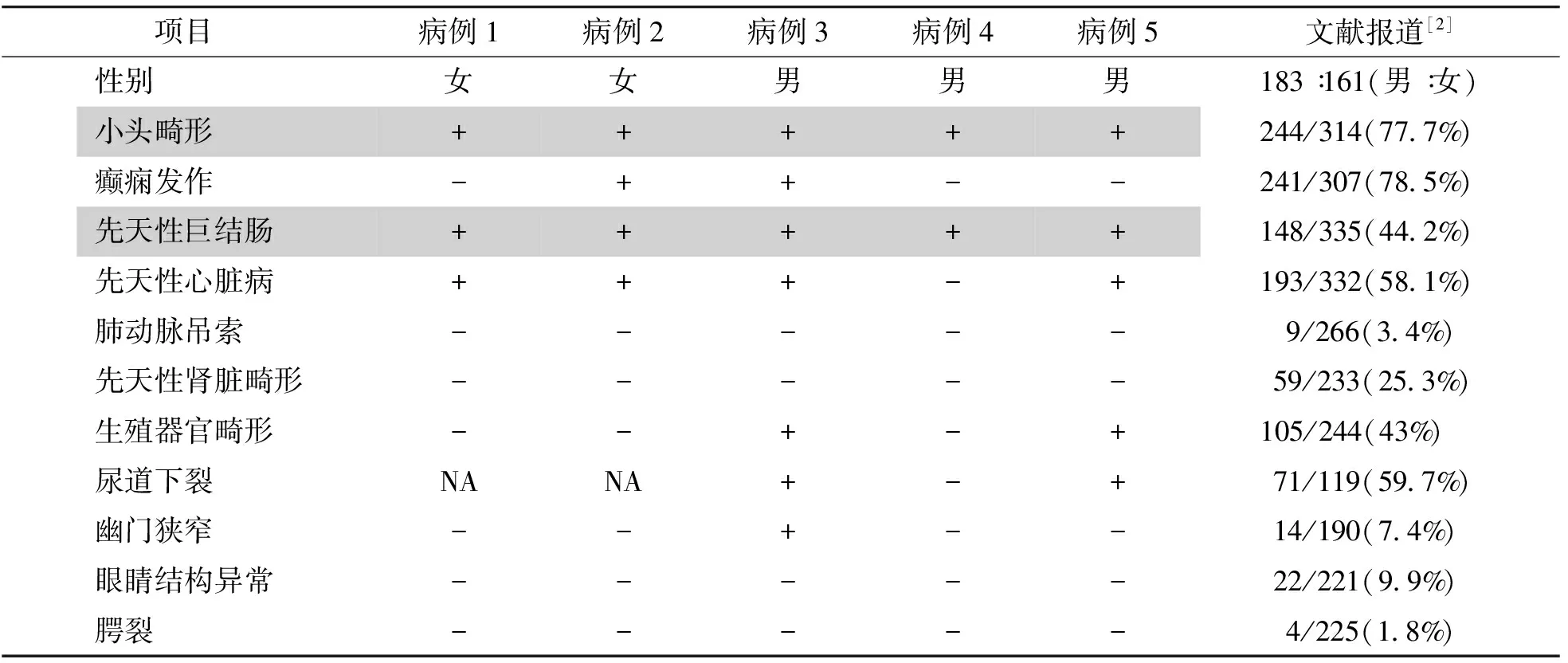
项目病例1病例2病例3病例4病例5 文献报道[2]性别女女男男男183∶161(男∶女)小头畸形+++++244/314(77.7%)癫痫发作-++--241/307(78.5%)先天性巨结肠+++++148/335(44.2%)先天性心脏病+++-+193/332(58.1%)肺动脉吊索----- 9/266(3.4%)先天性肾脏畸形----- 59/233(25.3%)生殖器官畸形--+-+105/244(43%)尿道下裂NANA+-+ 71/119(59.7%)幽门狭窄--+-- 14/190(7.4%)眼睛结构异常----- 22/221(9.9%)腭裂----- 4/225(1.8%)
NA表示女性患儿不考虑该项异常;本研究中全部5例患儿均存在的症状用灰色突出显示;“/”前后的数字分别代表有该类症状的患者数及文献中相关信息可查询的患者总数,括号中为MWS患者中具有该项症状者所占的百分比
2.2 ZEB2基因检测结果
5例患儿均携带ZEB2基因新发杂合突变,其中3例为无义突变(病例1:c.904C>T,[p.Arg302X];病例2:c.756C>A,[p.Tyr252 X];病例3:c.2761C>T,[p.Arg921X]),2例为移码突变(病例4:c.264-267delAAGG,[p.Ile88Metfs*18];病例5:c.211delT,[p.Ser70Profs*3])。3例患者(病例1,3,4)携带的为已知致病突变,2例(病例2,5)为未报道过的新变异(p.Tyr252 X、p.Ser70Profs*3,见图2)。
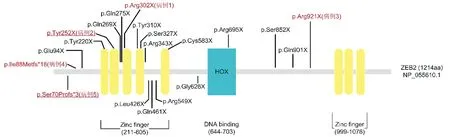
Zinc finger代表锌指功能域;HOX代表DNA结合功能域。既往文献报道的MWS患者无义突变(数据来自ClinVar:https://www.ncbi.nlm.nih.gov/clinvar)用黑色字体表示,本研究确诊的5例患者突变用红色字体表示,新发现的2个突变用下划线标记
2.3 新变异致病性分析
病例2携带ZEB2基因c.756C>A(p.Tyr252 X)杂合变异,病例5携带ZEB2基因c.211delT(p.Ser70Profs*3)杂合变异,Sanger测序证实两位患儿的父母均未携带相应变异,该患儿为新发变异。两例患儿均无家族史,携带的变异均属于无功能变异(null variants),且为在ESP数据库、千人基因组数据库、ExAC数据库正常对照人群中均未发现的变异,根据PVS1+PS2+PM2的联合判定标准[8],可判定为“致病的”变异。
3 讨论
MWS是一种具有高度表型和遗传异质性的先天发育异常类疾病,目前国际上尚无明确的诊断标准,给早期临床诊断带来巨大困难。本病临床表现多样且个体差异较大,与多种综合征都存在表型交叉[9,10];唯一可作为拟诊提示性证据的患儿的独特面容又往往在婴幼儿时期不甚典型,极易造成漏诊、误诊[11]。近年来,随着医学遗传学尤其是分子遗传学的快速发展,现代疾病的诊疗模式发生了巨大变化,发展为基于临床诊断-影像学诊断-基因诊断的立体模式[12],对于临床可疑患者,应及时进行相关检测,争取实现早期诊断和早期干预。包含所有HSCR相关基因的基因panel检测是一种经济、高效、快速的检测手段,可为临床医生在第一时间提供所有相关基因突变的整体情况,为精准的临床诊断和后期随访提供可靠保障[13]。
ZEB2基因又称锌指同源框基因1B(zinc finger homeobox gene 1b,ZFHX1B)或Smad交互蛋白1(Smad interacting protein 1,SIP1),是神经系统发育过程中的多功能调节因子[14],其编码蛋白的表达贯穿于神经元及神经胶质细胞的整个生长发育过程,是参与神经系统发育调控的重要转录因子[15]。目前已报道的典型MWS患儿的致病原因主要包括ZEB2基因的大片段缺失、移码突变和无义突变[16]。其中无义突变在整个基因编码蛋白的各个功能域中散在分布,无明显热点突变[17]。迄今为止,共有4个无义突变曾在不同种族的患者中多次出现,分别是:c.904C>T(p.Arg302X)、c.1027C>T(p.Arg343X)、c.2083C>T(p.Arg695X)及c.2761C>T(p.Arg921X),基因型相同的患者呈现出的临床表型却不尽相同,提示存在较大的遗传异质性[12,18]。本研究共确诊5例MWS患者,全部由点突变引起。其中2例患者的致病突变既往均未见报道,对于扩展该类疾病的基因突变谱具有一定意义。虽然全部患者都以先天性巨结肠为首诊症状,但合并的其他畸形各异;病例3携带的无义突变最靠近编码蛋白的C-末端,但该患者的临床表型却显著重于突变位置比较靠前的病例2和病例4,与既往文献报道一致。
除面容异常外,全部确诊的MWS患者中发生频率最高的5个症状依次为:癫痫(78.5%)、小头畸形(77.7%)、尿道下裂(59.7%)、先天性心脏病(58.1%)及身材矮小(46.4%)[2],可为临床医生提供一定诊断线索。绝大多数患者都是由新发突变引起,但有少数家族性患者可能是由父母一方的生殖腺嵌合导致的[19,20]。鉴于孕期B超检查可能受限于观察角度和胎儿肢体的阻挡,且多数异常征象亦可见于其他先天发育异常类疾病,特异性不强,故大部分MWS患儿都是在生后才被确诊。Wilson等[21]回顾了2例确诊MWS患儿的孕期检查结果,发现除颈部半透明带增宽外其余均未有明显异常,提示遗传咨询在降低本病的发生率上具有至关重要的作用,应予以高度重视。
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2020年5期)2021-01-14
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
百科知识(2015年18期)2015-09-10
中国当代医药(2015年30期)2015-03-01
