
基于WGP物理作图法的虾夷扇贝Fosmid克隆混池策略及解码率研究*
2019-12-07 12:52窦怀乾李仰平窦锦壮李语丽
中国海洋大学学报(自然科学版) 2019年2期
窦怀乾, 李仰平, 吕 佳, 窦锦壮, 李语丽, 王 师,2**
(1.中国海洋大学海洋生物遗传学与育种教育部重点实验室,山东 青岛 266003;2.海洋生物学与技术功能实验室,青岛海洋科学与技术国家实验室,山东 青岛 266237)
虾夷扇贝(Patinopectenyessoensis)于1980年代初由日本引入中国[1],并在山东、辽宁等北方沿海地区进行大规模人工养殖。目前已在渤海及黄海北部形成规模化和产业化养殖,成为中国北方最重要的海水养殖贝类之一[2]。
近几年,随着二代高通量测序技术(Next generation sequencing, NGS)技术快速的发展,组学研究成为了当前的热点。目前,利用高通量测序技术已经完成了许多海洋软体动物基因组的测序和拼接,例如:马氏珠母贝(Pinctadafucata)[3]、太平洋牡蛎(Crassostreagigas)[4]、帽贝(Lottiagigantea)[5]、章鱼(Octopusbimaculoides)[6]、虾夷扇贝(Patinopectenyessoensis)[7]等。在基因组拼接的过程中,物理图谱是一个最重要的辅助完成全基因组拼接,提高基因组拼接水平的工具之一。物理图谱基于逐步克隆(Clone-by-clone)的策略[8]获得各个克隆的相对位置;其与基因组草图结合,能对基因组中的重复序列和拼接断点等区段进行修正和连接,不仅能显著提升拼接的效果,更能对拼接得到的基因组进行校正。
利用Fosmid大片段文库已完成多个物种物理图谱的构建,如:人(Homosapiens)[9]、大猩猩(Gorillagorilla)[10]、水稻(Oryzasativa)[11]、长臂猿(Nomascusleucogenys)[12]、甜菜(Betavulgaris)[13]等。这种方法在海洋生物中也有广泛的应用,例如海鞘(Botryllusschlosseri)[14],半滑舌鳎(Cynoglossussemilaevis)[15]、斑节对虾(Penaeusmonodon)[16],栉孔扇贝(Chlamysfarreri)[17]等。构建物理图谱的方法有SNaPshot技术[18]、BioNano[19]、OpticalMap[20]、WGP技术等[21],而WGP (Whole genome profiling,全基因组解析)物理图谱是一种基于大片段克隆文库混池策略及克隆特异序列标签的新型物理图谱构建方法。它利用克隆间标签序列的特异性来确定克隆的重叠关系,排除了DNA指纹技术酶切条带大小差异产生的误差,提高了精度和可靠性;与高通量测序结合,不需要对酶切片段进行电泳,极大简化了实验操作步骤。利用WGP法,van Oeveren等完成了拟南芥(Arabidopsisthaliana)物理图谱[21]的构建。以拟南芥BAC文库6 144个克隆为材料,采用EcoRI/MseI两种限制性内切酶获取标签,最终解码4 599个克隆,克隆解码率为74.8%;构建完成的物理图谱实现了98%的标签准确排序,覆盖了97%的基因组。Nicolas Sierro等利用WGP法构建了烟草的物理图谱[22],对425 088个BAC克隆,采用EcoRI/MseI两种限制性内切酶获取标签,成功解码361 034个,解码率84.9%。
在利用WGP法进行物理图谱构建时,实验中的克隆混池策略,即多少张板(克隆)混合在一起,是物理图谱构建的关键。在选定的混合尺度下,既需要控制混合的克隆数目,保证能够获得高克隆解码率,又需要能尽可能多的混合克隆,来平衡测序的成本。因此,在进行WGP法物理图谱构建之前,对克隆混池策略及解码率进行研究,就显得尤为重要。本实验室于近期完成了虾夷扇贝基因组图谱[7]的绘制工作,但基因组的高复杂度及高重复序列含量,使得某些区段会产生拼接的错误,物理图谱便可以对这些错误的拼接进行校正[23]。因此本研究基于构建完成的虾夷扇贝Fosmid大片段文库,对4、8、12、16张384孔培养板混合尺度下Fosmid克隆解码率进行了分析,为后期采用WGP法构建虾夷扇贝物理图谱提供了混池和解码方案的参考。
1 材料与方法
1.1 Fosmid文库的构建
1.1.1 实验材料 实验材料为新鲜虾夷扇贝活体解剖后,保存于-80 ℃超低温冰箱的肉柱(横纹肌)。试剂盒为Epicentre CopyControlTMFosmid 文库构建试剂盒。
1.1.2 虾夷扇贝基因组DNA的提取及末端修复 虾夷扇贝的DNA采用酚/氯仿抽提法提取,使用1%琼脂糖凝胶电泳检测DNA完整性,并利用核酸蛋白质检测仪(NanoVue spectrophotometer, General Electric)检测A260/A280、A260/A230以及DNA浓度,确保样品合格。然后对基因组DNA进行末端修复,以保证片段和载体连接效率。反应的组分如下:20 μg DNA x μL,End-Repair 10×Buffer 8 μL,0.25 mol/L dNTPs 8 μL,1 mmol/L ATP 8 μL,End-Repair Enzyme Mix 4 μL,ddH2O (52-x) μL。22 ℃恒温45 min,加入Loading Buffer后,70 ℃,灭活10 min。
1.1.3 目的片段回收及目的片段与载体连接 末端修复的产物,经脉冲场凝胶电泳进行目的片段的回收,具体方法如下:采用线性自动程序,20~60 kb输入片段大小,0.5×TBE电泳液,电泳16 h;EB染色10 min,根据Marker位置,切取40 kb大小的目的片段,经Epicentre CopyControlTMFosmid试剂盒进行目的片段的回收;然后将目的片段和试剂盒中提供的载体(CopyControl pCC1FOSTM)进行连接。
连接反应10 μL,体系如下:0.25 μg插入片段6 μL,10×Fast-Link连接缓冲液1 μL,10 mmol/L ATP 1 μL,pCC1FOS 载体(0.5 μg/μL)1 μL,Fast-Link DNA连接酶(试剂盒内置)1 μL。置于PCR仪(Bio-Rad),22 ℃恒温2 h;70 ℃灭活10 min。
1.1.4 质粒包装与噬菌体转导 将连接产物与试剂盒中的MaxPlax Lambda Packaging Extracts包装蛋白进行包装,每次25 μL,连续包装2次。待包装完成之后,补加25 μL氯仿和940 μL噬菌体稀释缓冲液(PDB)混匀,然后取10 μL混合样与提前准备好的A600在0.8~1.0的宿主菌(试剂盒提供)100 μL进行噬菌体的转导,可同时转导多个样品。
1.1.5 克隆的培养及获取 将多个上述110 μL混合体系分别涂布到LB固体平板(氯霉素12.5 μg/mL)进行克隆培养。在克隆生长到合适大小,利用灭菌牙签将克隆逐一挑取到无菌且添加了400 μL(氯霉素12.5 μg/mL)液态LB的96孔深孔板中。37 ℃过夜培养,然后添加甘油,至终浓度为20%,混匀之后转移到384孔板中保存。至此,即完成虾夷扇贝Fosmid文库的构建。
1.2 数据的模拟分析
在WGP测序文库构建之前,以实验室的虾夷扇贝基因组数据为基础,利用计算机进行混合克隆数目的模拟。具体方式如下:虾夷扇贝基因组大小约1 Gb,用BsaXI和FspEI两种酶进行酶切;从基因组中随机获得384N(N=1,2…24)个40 kb片段,一个片段即代表一个克隆,模拟克隆在384孔培养板中的分布分别进行行池、列池、板池的编号;获得相应倍数下,片段内的BsaXI和FspEI两种酶酶切标签总和,并统计标签唯一位置出现的概率,获得混池策略的模拟结果。
在具体的实验中,需要在384培养板混合数目和解码率两者之间进行取舍。更少的板数混合会获得更高的解码率,但实验通量较小,单一混合样本包含的克隆数目较少,为了获得特定的覆盖度,测序的成本会增加。反之,更多的板数混合虽然会带来解码率的降低,但能在单一样本的测序中包含更多的克隆数目,降低测序的成本。所以实验中需要选择合适的混合尺度来平衡解码率和混合尺度相关连的测序成本。
1.3 WGP测序文库的构建
1.3.1 实验材料 实验材料为构建完成的虾夷扇贝Fosmid文库克隆:保存于-80 ℃超低温冰箱,共计320张384孔培养板,122 880个克隆。实验开始前,挑取384孔板中的克隆置于96孔培养板中进行活化。
实验中对超级池,行池,列池的界定如下。以8张为例:超级池:8 张 384 孔培养板的克隆。二级池:8 张 384 孔培养板叠在一起的行和列的克隆,称行池和列池(16行24列)。板池:每张 384 孔培养板对应的克隆。这样每 8 张 384 孔培养板中,就包括了 1 个超级池,16 个行池,24 个列池,8 个板池,共 49 个池。该研究构建了4、8、12、16张384 孔培养板为单位的4个混合尺度共200个池。
1.3.2 质粒提取 用碱裂解法[24]提取通过混合克隆获得的每个行池、列池、板池的质粒DNA(共200个样品),使用1 %琼脂糖凝胶电泳检测质粒DNA完整性,并利用核酸蛋白质检测仪检测A260/A280、A260/A230以及DNA浓度,确保样品合格。然后统一稀释至200 ng/μL,以便后续实验使用。
1.3.3 2b-RAD测序文库构建 将提取的Fosmid克隆质粒进行2b-RAD测序文库的构建[25]。本研究采用了限制型内切酶BsaXI,和修饰依赖型内切酶FspEI两种酶,进行固定标签长度的2b-RAD型文库的构建。
(1) 限制性内切酶酶切
BsaXI(New England Biolabs)酶切体系15 μL,组成如下:质粒DNA 1 μL,Buffer 4 1.5 μL,BsaXI 2 μL,ddH2O 11.5 μL。设置酶切反应空白对照组,内切酶用同等体积 ddH2O 替代,其余组分同实验组一致。37 ℃恒温孵育3 h。取2 μL样品,1%琼脂糖检测DNA条带是否被切开。
FspEI酶在酶切之前,需要利用M.SssI酶对质粒DNA进行预处理,使其所有CG位点全部转化成可被FspEI识别的甲基化酶切位点。具体反应体如下:M.SssI(New England Biolabs)反应体系20 μL,组成如下:质粒DNA 1 μL,10×Buffer-2 2 μL,10×SAM 2 μL,M.SssI 1 μL,ddH2O 14 μL。37 ℃恒温孵育1 h,65 ℃灭活20 min。
FspEI(New England Biolabs)酶切体系30μL,组成如下:M.SssI处理产物10 μL,10×CutSmart Buffer 3 μL,30×Enzyme Activator Solution 1 μL,FspEI 1 μL,ddH2O 15 μL。设置酶切反应空白对照组,内切酶用同等体积 ddH2O 替代,其余组分同实验组一致。37 ℃恒温3 h, 80 ℃灭活20 min。取2 μL样品,1%琼脂糖检测DNA条带是否被切开。
(2) 接头连接
利用T4 DNA连接酶在BsaXI酶切标签两端连接Illumina 测序平台特异接头Ad1(Slx-MpAd1-NNN)和Ad2(Slx-MpAd2-NNN)[25];而FspEI标签两端连接的接头为Ad1(Slx-MpAd1-NNNN)和Ad2(Slx-MpAd2-NNNN)。
T4连接反应体系22 μL,组成如下:酶切产物DNA 10 μL,ATP (10 mmol/L) 2 μL,Ad1(5 μmol/L) 0.8 μL,Ad2(5 μmol/L) 0.8 μL,T4 ligase buffer 2.2 μL,T4 ligase 2 μL。4 ℃恒温过夜。
(3) 1stPCR扩增
用Phusion高保真DNA聚合酶(New England Biolabs)和Illumina测序平台特定引物1stP1(Slx-1st-MpPrimer-1) 和1stP2(Slx-1st-MpPrimer-2)对连接过接头的标签进行一轮PCR扩增,来增加标签的数目。
1stPCR扩增体系20 μL,组成如下:连接产物7 μL,5×H buffer 4 μL,dNTPs(10 mmol/L) 0.6 μL,1stP1(10 mmol/L)0.4 μL,1stP2(10 mmol/L) 0.4 μL,Phusion DNA Polymerases 0.2 μL,ddH2O 7.4 μL。PCR条件如下:(98 ℃ 5 s, 60 ℃ 20 s, 72 ℃ 10 s),14个循环,72 ℃延伸10 min。
(4) 1stPCR产物回收
1stPCR产物经10%非变性聚丙烯酰胺凝胶300 V/60 min电泳后,回收约100 bp的目标片段。充分研磨胶块,加入40 μL ddH2O,4 ℃透析过夜。天根CA2柱过滤回收获得1stPCR产物。
(5) 2ndPCR扩增
提纯后的1stPCR产物与二轮扩增引物 2ndP1(Slx-2nd-MpPrimer)和2ndP2(Slx-index-Barcode进行第二轮PCR扩增,用来引入Barcode序列。(2ndP2中含有一段Barcode序列,能够在平行测序中区分来自不同样品的标签)。
2ndPCR扩增体系20 μL,组成如下:1stPCR产物3 μL,5×H buffer 4 μL,dNTPs(10 mmol/L) 0.6 μL,2ndP1(10 mmol/L) 0.2 μL,2ndP2(10 mmol/L) 0.2 μL,Phusion DNA Polymerases 0.2 μL,ddH2O 11.8 μL。PCR条件如下:(98 ℃ 5 s, 60 ℃ 20 s, 72 ℃ 10 s),7个循环,72 ℃延伸10 min。
(6)产物纯化
采用QIAquick PCR Purification Kit (QIAGEN) 纯化试剂盒纯化PCR产物,35 μL的ddH2O洗脱DNA。
(7)文库测序
构建完成的2b-RAD测序文库,采用Illumina HiSeq 2000 平台进行单末端测序(36SE)。
1.3.4 测序数据的分析 测序获得的数据利用本实验室开发的RADtyping[26]软件进行分析,详细步骤如下:
(1)质量过滤:将原始测序文件中,含N及碱基平均质量低于30的序列去除。
(2)文库划分:根据序列中添加的Barcode(2ndP2),将来自相同行池,列池的标签分到一起。
(3)提取BsaXI和FspEI标签:根据两种酶的酶切位点提取酶切标签。
(4)去除污染标签:将提取到的酶切标签分别与宿主细菌基因组序列和载体序列比对,并去除比对上的污染标签。
(5)标签聚类:对得到的标签无错配的聚类,只保留出现大于等于2次的标签,去除可能测序错误数据。
(6) 解码克隆:根据1.3.1中的混池策略,解码标签(克隆)。解码规则如下:a在对应的行池、列池、板池中,只出现一次的标签,意味着它在三维坐标中拥有唯一的点,能够定位到单个克隆。标签对应的克隆即为解码出的克隆,这样克隆能够用于重叠群的建立。b在行、列、板三种池中都出现,但二级池出现多次的标签,能够定位到多个克隆。c只出现在1种或2种二级池的标签,无法定位到克隆。定位到多个克隆及无法定位到克隆的标签,都无法用于后续克隆重叠群的建立。
通过上述解码分析,统计解码出的克隆个数,获得解码率。并统计每个超级池中单个克隆解出的标签数及标签测序深度结果。
2 结果
2.1 Fosmid文库构建
2.1.1 目的片段的回收 在文库的构建中,目的片段的回收至关重要,回收片段的质量决定了后期连接的效率。该实验中,实验获得的目的DNA具有很高的片段一致性(见图1)。
2.1.2 转导效果 10 μL包装产物转导涂板后,每张平板(直径15 cm)一般可形成约1 000个克隆左右,效价为1×105cfu/mL。图2所示为包装产物的转导结果图,克隆数目较多,大小较为一致,转导效果好良好。
2.2 WGP测序文库的分析
2.2.1 模拟数据结果分析 混合克隆数目的数据模拟分析结果见图3。
由图3可知,随着混合的384孔板的数目的增加,即克隆数目的增加,解码率呈现逐渐下降的趋势。BsaXI对应的解码率从最开始的99.74%下降到85.66%,FspEI对应的解码率从最开始的99.48%下降到93.29%,前者比后者下降的幅度更大。

(M1:λ DNAHindIII digest;M2:42 kb Fosmid control DNA;1~2:切胶回收的目的DNA片段。M1:λ DNAHindIII digest; M2:42 kb Fosmid control DNA; 1~2: DNA fragments.)
图1 脉冲场凝胶电泳检测切胶回收DNA片段
Fig.1 DNA fragments detection with Pulse field gel electrophoresis
图2 噬菌体转导克隆培养结果图
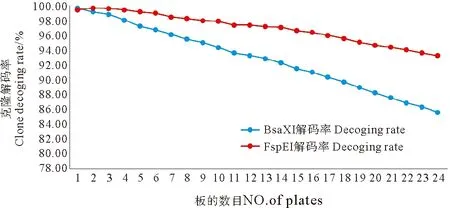
图3 超级池构建策略的模拟结果
综合模拟数据的结果以及实验成本的因素,最终挑选了模拟混合尺度中的4、8、12、16这4个混合尺度去进行实验部分的超级池及对应测序文库的构建。
2.2.2 测序数据统计 本研究中以BsaXI和FspEI两种限制性内切酶构建的2b-RAD文库,经Illumina HiSeq 2000测序后,获得的原始测序reads 经过质量过滤,得到高质量reads。如表 1所示,高质量reads均占测序原始reads的99%以上,测序数据质量较高,可用于后续分析。

表1 测序数据统计
2.2.3 标签统计 以8张384孔培养板板混合得到的二级池为例。21 356个BsaXI酶切标签中,定位到单个克隆的标签数为1 054,占总标签数的47.08%;定位到多个克隆的标签数为5 063,占总标签数的23.71%;无法定位到克隆的标签数为6 239,占总标签数的29.21%。141 166个FspEI酶切标签中,共有40 553个标签成功定位到单个克隆,占总标签数的28.73%;定位到多个克隆的标签数为26 046,占总标签数的18.45%;无法定位到克隆的标签数为74 567,占总标签数的52.82%。其他混合条件下的结果如表2所示。整体来说,约有50%的BsaXI酶切标签中能定位到克隆上;约有30%FspEI酶切标签能定位到克隆上。
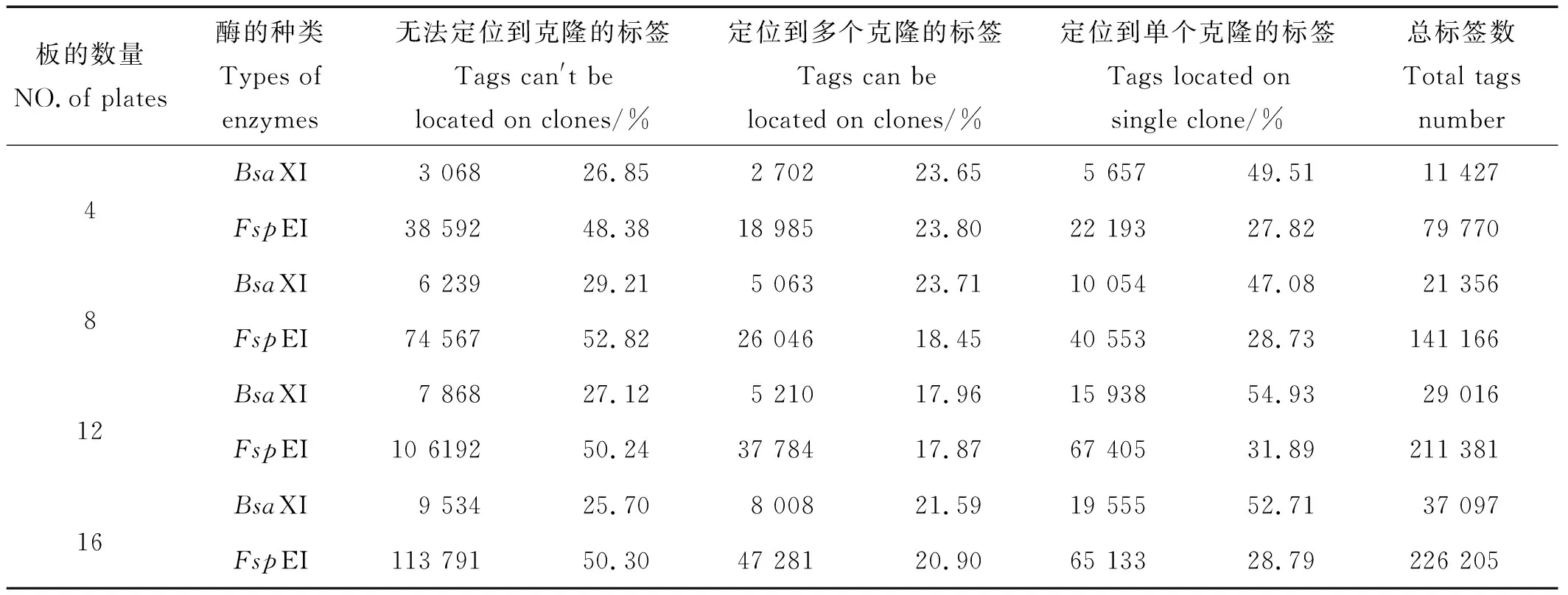
表2 BsaXI和FspEI酶切标签解码统计
2.2.4 克隆解码率和平均标签数统计 以8张384孔培养板混合得到的二级池为例,此超级池共包含克隆3 072(8×16×24)个。10 054个定位到克隆上的BsaXI酶切标签,成功解出克隆2 179个,克隆的解码率为70.93%,平均每个克隆所含的标签数为4.61。40 533个定位到克隆上的FspEI酶切标签,成功解出2 668个克隆,克隆解码率为86.85%,平均每个克隆所含的标签数为15.2。联合分析BsaXI和FspEI两种酶切标签,克隆解码率为88.41%。
其他混合尺度下解码情况如表3所示,随着超级池所含的384孔培养板板数的增加,即克隆数量的增加,2种酶的解码率都明显下降。4种尺度下,FspEI酶对应的克隆平均标签数在15个左右,BsaXI酶对应的克隆平均标签数在5个左右。FspEI酶的克隆解码率高于BsaXI酶的解码率;联合两种酶切标签进行解码,克隆解码效率得到明显提升。
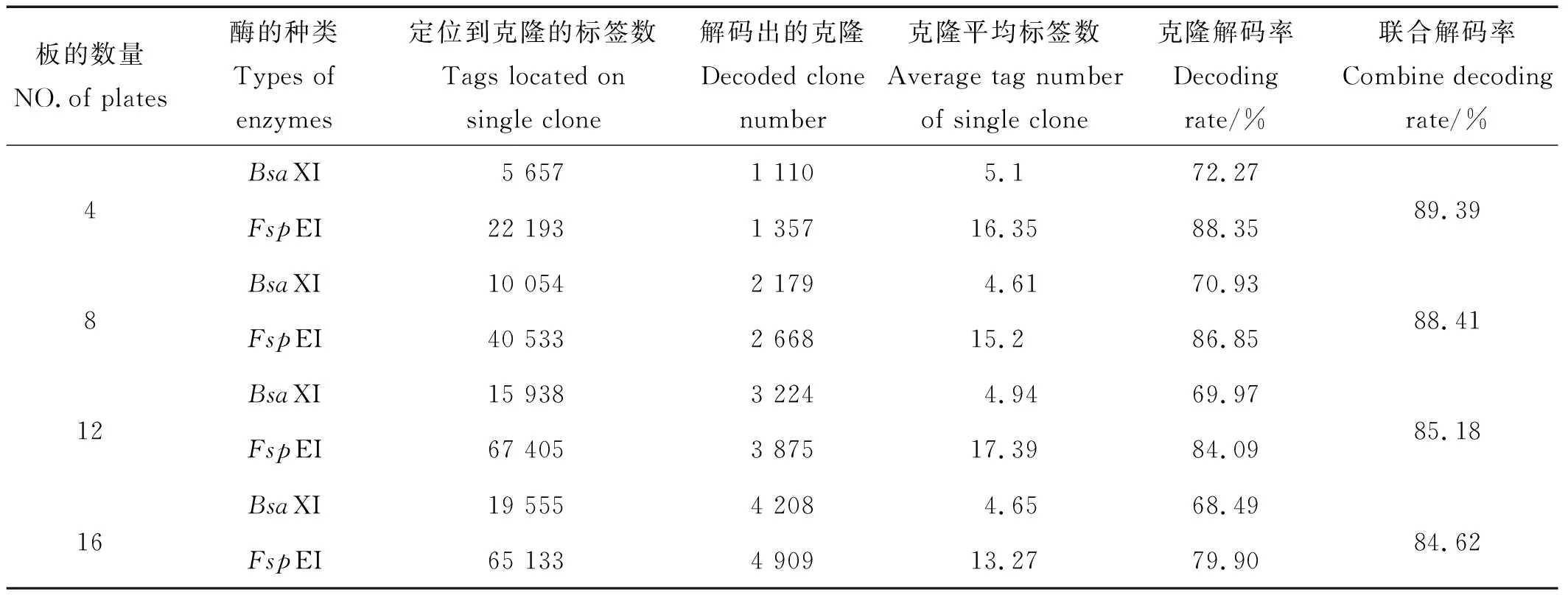
表3 不同混合尺度下克隆的解码率
3 讨论
本研究中,测序得到的酶切标签根据解码情况可以分为三类。在对应的行池、列池、板池中,只出现一次的标签,意味着它在三维坐标中拥有唯一的点,能够定位到克隆上,这样的克隆解码出的克隆。这样的标签(克隆)能够用于重叠群的建立。在行、列、板三种池都出现,但二级池出现3次及3次以上的标签,能够定位到多个克隆。只出现在1种或2种二级池的标签,无法定位到克隆。后两者都不能用于后续重叠群的分析。对于定位到多个克隆的标签,说明这个标签出现在基因组的多个位置,即该标签有可能是是重复序列的一部分。对于无法定位到克隆的标签,可能是因为克隆生长不均匀,导致在取样时有的二级池没有取到含有该标签的克隆(例如仅在行池和列池中检测到,而未在板池中检测到)。
对于定位到克隆的两种酶切标签的比例,BsaXI的标签有效率约为50%,FspEI的标签有效率约为30%。这可能是由于FspEI为4碱基内切酶,在虾夷扇贝基因组中有比BsaXI更多的酶切位点,相同的混合尺度下,重复序列来源的标签更容易被测到;相同的测序深度下,定位到克隆的标签更不容易被测到。同一种酶切标签,混合克隆的数量提高解码率会跟着降低。从4个384孔培养板板增加到16个,BsaXI酶的解码率由72.27%降到68.49%,FspEI酶的解码率由88.35%降到79.90%。对比两种酶切标签,定位到克隆的FspEI标签数明显多于BsaXI标签数,FspEI酶切标签的克隆解码率明显高于BsaXI酶切标签的克隆解码率。
此外,可以通过构建更长片段的克隆文库(如BAC文库),来提高定位到克隆的标签数,获得更高的解码率。更长的克隆片段包含更多的酶切位点,每个克隆中所能检测到的标签数目也会越多,克隆被解码的概率也随之增大。克隆片段长度的增加,组成克隆重叠群的克隆数目会越少,也更有利于克隆之间的正确排序。达到相同的基因覆盖度,更长片段的克隆文库需要更少的克隆进行混池,也能降低WGP法混合池构建的成本及测序的成本;但插入片段越长,构建克隆文库的难度越大,实验成本也会随之增加,需要在文库的插入片段长度、成本和实验难度上做权衡。
综合以上分析,在后期的实验中同时使用两种酶,能够使每个克隆所含的标签数目提高,获得比一种酶更高的解码率。在结合实验成本,测序成本及比对实验的4个混合尺度下的综合解码率之后,本研究最终确定了由8张384孔培养板混合的混池策略及利用BsaXI和FspEI两种酶切标签联合进行克隆解码的解码方案。该方案联合解码率达到88.41%,比逐个克隆进行测序解码率更高;在获得高解码率的同时,单个混合样品包含更多的克隆,降低了测序成本,为后期利用WGP方法构建虾夷扇贝物理图谱,提供了克隆混池和解码参考方案。
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
中国石油石化(2022年12期)2022-07-16
猪业科学(2021年3期)2021-05-21
幽默大师(2020年10期)2020-11-10
家教世界·创新阅读(2019年11期)2019-12-10
中国外汇(2019年19期)2019-11-26
中华诗词(2019年1期)2019-11-14
红领巾·探索(2019年8期)2019-08-21
家庭影院技术(2018年11期)2019-01-21
家庭影院技术(2018年11期)2019-01-21
