
Co、Ni和Ga合金化对DO19-Ti3Al弹性性能影响的第一性原理研究
2019-12-06 09:31:48张超彦张丽霞
原子与分子物理学报 2019年6期
张超彦 , 张丽霞 , 侯 华
(1. 徐州工业职业技术学院,徐州 221100; 2. 中北大学,太原 030051)
1 引 言
在可持续发展战略下,航空及汽车等行业广泛关注轻量化技术,对材料的比强度要求日益增加[1]. 全层片状TiAl合金具有良好的高温强度、比强度及比刚度等特点在轻量化设计中潜力巨大,研究者在成分设计、表面处理和加工方式等领域开展了广泛的研究. 截至目前,室温塑性较低依然制约其应用范围,室温塑性差的主要原因为γ相中的电荷方向性较强及α2相变形中晶界应力由于滑移系较少不能释放产生脆性断裂[2, 3]. 合金化被广泛应用于TiAl合金的性能改善,对γ相,合金元素加入可以消弱其晶体内的电荷方向性从而改善其塑性;对α2相则主要是降低Ti-Ti键的强度从而改善其非基面滑移的阻力[4, 5].
随着计算材料机技术的发展,采用计算方法研究材料的性能越来越普及,第一性原理研究被广泛应用于TiAl合金. Liu[6]等采用第一性原理对α2-Ti3Al晶体的电子结构和弹性性能进行研究,并解释了其变形机制;Music等[7, 8]研究了V、Nb和Ta对γ-TiAl晶体化学键的影响,发现这些元素可以改善其弹性性能,并且对V、Nb、Ta、Cr、Mo、W和Mn元素在α2相中的固溶体进行计算,得出其弹性常数并分析其电子结构,发现合金元素可以降低晶体中的共价键强度,从而提高合金的塑性;王海燕[9]通过不同浓度的固溶体模型研究了Mo元素浓度对γ-TiAl合金的性能影响,发现Mo元素浓度为12.5at.%可以显著改善合金的塑性;Zhang等[10]的第一性研究发现Nb和Ta元素固溶降低α2-Ti3Al的塑性,但可以显著提高其硬度.
合金元素对层片状TiAl合金中主相的性能影响的研究已广泛开展,但主要集中在γ相中,对于α2相的研究相对薄弱,合金元素种类也较少,截至目前未见Co、Ni和Ga元素对Do19结构的α2-Ti3Al的弹性性能和电子结构的相关报导. 由于Co元素做为常见的合金元素广泛应用于高硬度合金和高温合金,Ni元素做为合金元素通常改善材料的塑性及抗腐蚀性,Ga元素置换Al元素可以改善NiAl基合金的室温塑性,故对此3种元素对Do19-Ti3Al的性能影响研究,为实验提供理论基础.
2 计算模型与方法
DO19-Ti3Al的晶体结构为密排六方结构,空间群No.194,其单胞模型由6个Ti原子和2个Al原子组成,原子坐标分别为(1/6,5/6,3/4) 和(1/3,2/3,1/4). 由于Ti和Al原子与M(M=Co、Ni和Ga)原子的半径之比分别为1.05、1.06、0.94和1.13、1.15、1.0,均大于0.59,在Ti3Al晶体中M原子以置换原子的形式固溶[11]. 固溶体的计算模型采用1×1×2的Ti3Al超晶胞,其中包含12个Ti原子和4个Al原子,M原子置换其中的一个Ti原子(Al原子),得到浓度6.25 %的Ti11Al4M(Ti12Al3M)晶体,如图1所示.

图1 Ti11Al4M(Ti12Al3M)(M=Co、Ni和Ga)的晶体结构(其中:灰色球Ti原子,紫色球Al原子,红色球M原子)Fig. 1 The crystal structure of Ti11Al4M (Ti12Al3M) (M=Co, Ni and Ga)
采用基于密度泛函理论的第一性原理MS程序包中CASTEP模块,赝势选取为超软(Ultrasoft)赝势,计算在倒易空间中的进行,势能函数选择广义梯度近似的PBE和PW91两种形式进行对比. 平面波截断能Ecut和K点网格数的选取通过总能量收敛测试[12],分别确定为410.0 eV和6×6×3. 计算收敛条件设置为:能量波动小于5×10-6eV/atom,作用力小于0.01 eV/Å,应力小于0.02 GPa,位移小于5×10-4Å.
3 计算结果与分析
3.1 几何结构
通过对晶格的弛豫计算,得到Ti3Al晶体及其固溶体的平衡结构,计算所得的晶格参数和体积列于表1. 通过对表1中的计算数值与实验值和其它理论计算值的对比可以发现,采用势能函数PBE形式的平衡晶格常数误差更小,均在0.8%以内,故在后续的计算中选择PBE形式.
表1 Ti3Al的晶格常数(Å)和晶胞体积(Å3)
Tab.1 The lattice constant (in Å) and the cell volume (in Å3) of Ti3Al

体系来源晶格参数acc/a体积Ti3AlPBE5.7654.6570.807134.03-31.73PW915.7494.6540.810133.26Exp.[13]5.774.62--Exp.[14]5.77-0.80-Cal.[8]5.772-0.803-Cal.[15]5.744.65--
溶质原子取代溶剂原子得到的固溶体的形成焓可以反映其占位倾向,为此计算了固溶体的形成焓(ΔH),计算公式为:
(1)

Ti3Al晶体及其固溶体中M原子不同占位的形成焓ΔH计算值同样列于表1,可以看出,纯Ti3Al和M原子取代Ti(Al)的固溶体的形成焓均为负值,表明Co、Ni和Ga元素的置换固溶均放热,可以自发形成,且固溶体结构稳定不易分解. 比较形成焓的数值可以发现,Co和Ni原子占据Ti位时其形成焓较占据Al位小,Co和Ni更倾向于占据Ti;而Ga原子占据Al位时形成焓更小,Ga原子则倾向于占据Al位.

图2 Ti3Al-M合金体系的形成焓Fig. 2 The enthalpy of formation of the Ti3Al-M system
合金元素M固溶前后Ti3Al晶体的平衡晶格参数a、c及体积见表2,可知,Co、Ni和Ga元素在Ti3Al晶体中固溶均引起了平衡晶格常数和体积变小,原因为3种合金原子的半径均小于其置换原子,符合置换理论;密排六方结构的Ti3Al晶体c/a值约为0.807,Co、Ni和Ga元素固溶后合金体系的c/a值均发生变化,主要是由于合金原子半径的不同及成键特征的改变引起.
表2 Ti3Al-M(M=Co、Ni和Ga)的晶格常数(Å),晶胞体积(Å3)
Tab. 2 The lattice constant (in Å) and the cell volume (in Å3) of Ti3Al-M (M=Co, Ni and Ga)

体系晶格参数acc/a体积Ti3Al5.7654.6570.807134.03Ti11Al4Co5.7379.2141.606263.076Ti11Al4Ni5.7289.2141.608261.873Ti12Al3Ga5.7479.3201.622266.739
3.2 电子结构
材料的力学性能与其内部的电荷分布和原子间的成键特征等联系紧密[16],本文对Ti3Al晶体及其固溶体的电子结构进行了计算分析.
合金体系的总态密度图(DOS)见图3,图中虚线表示费米能级(设置为0),由图3可知,在纯净的Ti3Al晶体中,费米能级附近的成键峰分布在0.8 eV、-0.5 eV和-1.8 eV附近,其中0.8 eV和-0.5 eV附近的成键峰为Ti(d)-Ti(d)键,而在-1.8 eV附近的成键峰则来自杂化键Ti(d)-Al(p). Co、Ni和Ga元素在Ti3Al晶体中固溶均未明显改变总态密度形状,说明合金体系的基本物性与纯净Ti3Al保持一致,但可以发现,Co和Ni元素固溶后合金体系在0.8 eV、-0.5 eV和-1.8 eV附近的三个成键峰趋于平缓,可以推测Co和Ni元素的固溶能够削弱三个成键峰的电子杂化效应,降低成键强度;而Ga元素的固溶后成键峰变化不大.

图3 Ti3Al-M合金体系的态密度(M=Co、Ni和Ga)Fig. 3 The density of states of Ti3Al-M (M=Co, Ni and Ga) alloys
为了更直观的了解晶体内部的成键情况,绘制了Ti3Al晶体固溶前后的差分电荷密度图,如图4所示,电荷范围为-0.1至0.1 e/Å3. 由图4可知,纯净的Ti3Al中,层片间的Ti-Al-Ti原子中Ti原子的周围出现高密度电荷区域,其中间区域出现少量电荷聚集,表明层片间Ti-Al-Ti之间的成键主要为共价键和弱金属键;层片间Al-Ti-Al三原子中间区域电荷大量聚集,成键主要形式为强金属键;而层内的三个Ti原子附近均出现高密度电荷区域,三者之间的成键以强共价键形式为主. Co和Ni元素取代Ti原子后,Al-Co(Ni)-Al三原子之间的电荷聚集密度较Al-Ti-Al则明显降低,表明Co和Ni原子与Al原子之间的金属键强度下降,其中Co元素的作用更加明显;Ga原子取代Al原子后,Ti-Ga-Ti三原子间成键的强度较Ti-Al-Ti变化不大,原因为Ga原子与Al原子具有相同的成键电子数.
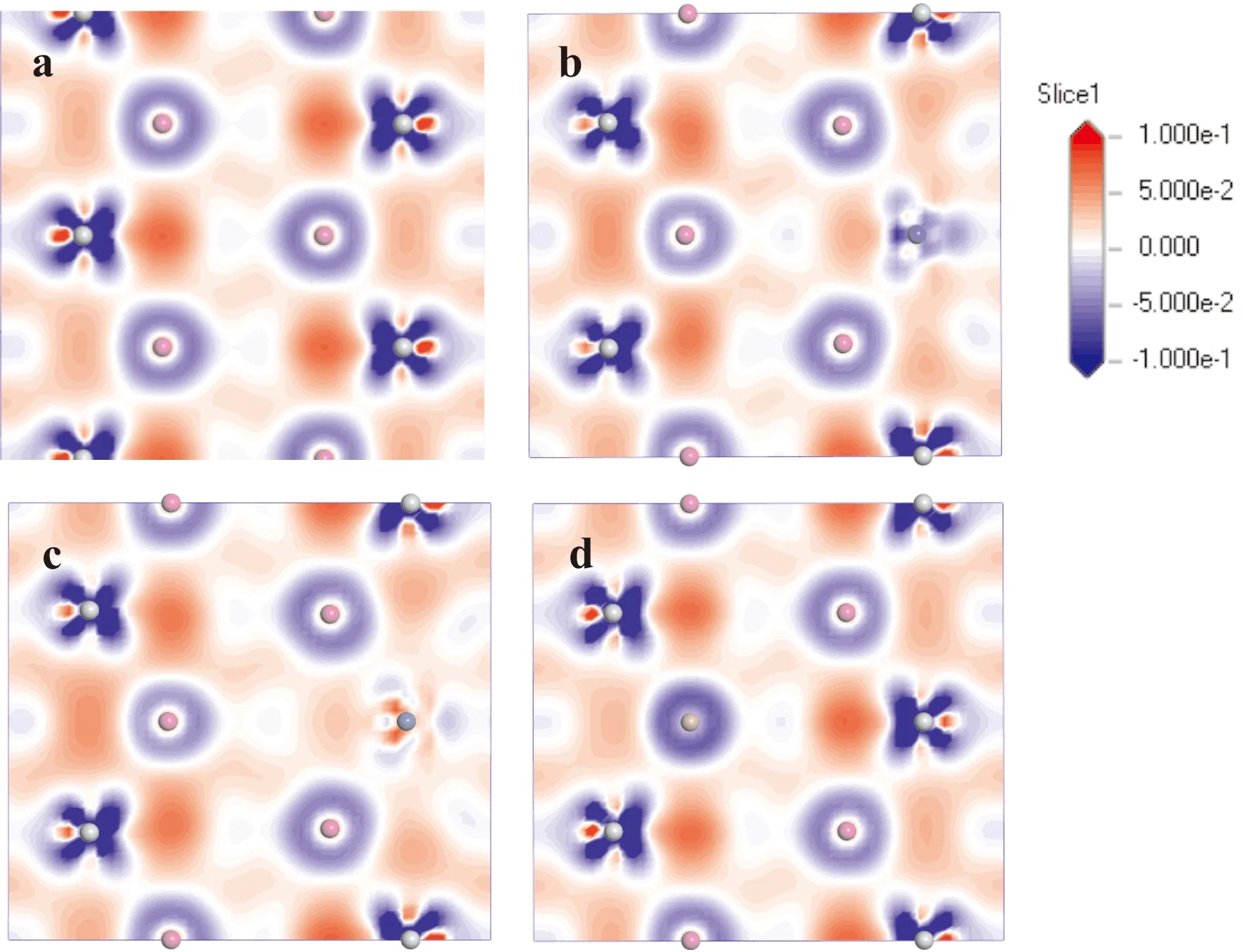
图4 Ti3Al-M晶体(110)晶面的电荷差分密度(a. pure; b. M= Co; c. M=Ni; d. M=Ga)Fig.4 The charge density density of the (110) plane on the Ti3Al-M crystal(a. pure; b. M= Co; c. M=Ni; d. M=Ga)
3.3 弹性性能
通过晶体的弹性常数的计算可以得到表征材料宏观力学性能的体模量、剪切模量及杨氏模量等性能指标[17],本文计算了Ti3Al晶体固溶前后的弹性常数列于表3. 合金原子的置换引起Ti3Al晶体的对称性改变,对于Ga元素取代Al原子后晶体仍为六方晶系,而Co和Ni原子取代Ti原子后晶体为正交晶系.
对于六方晶系和正交晶系,利用Voigt-Reuss-Hill方法可以通过其弹性常数计算得到体模量B、剪切模量G、杨氏模量E和泊松比υ,计算公式如下:
(2)
(3)

表3 Ti3Al-M晶体弹性常数(GPa)(M=Co、Ni和Ga)
(4)
(5)
(6)
对于六方晶系:
(7)
BR=C2/M
(8)
(9)
M=C11+C12+2C33-4C13
(10)
(11)

(12)
C66=(C11-C12)/2
(13)
对于正交晶系:
(14)
C33+3(C44+C55+C66)]
(15)
C23(C12C23-C23C11)+C33(C11C22-C122)
(16)
(17)
Δ=C11(C22+C33-2C23)+C22(C33-2C13)-
2C33C12+C12(2C23-C12)+
C13(2C12-C13)+C23(2C13-C23)
(18)
Δ1=C11(C22+C33+C23)+
C22(C33+C13)+C33C12-C12(C23+C12)-
C13(C12+C13)-C23(C13+C23)
(19)
合金体系的相关计算结果列于表4,可以看出:Co元素固溶后合金体系的体模量B增大,则合金体系的抗体积变形能力提升,而Ni和Ga元素固溶则导致体模量降低,其中Ni元素的降低作用更加明显;对于剪切模量G,Ga元素的提升效果明显,Co和Ni元素则降低作用明显;3种合金元素对晶体的杨氏模量E的影响与其对剪切模量G的影响相似,即Ga元素的固溶提升刚度,Co和Ni元素则降低刚度. 一般来说,材料的杨氏模量E和剪切模量G越大,其硬度越大,由表4可知,Ga元素固溶后合金体系的硬度将增大,而Co和Ni元素固溶将显著降低材料的硬度. 由于材料的弹性模量是由其晶体内原子间成键状态决定,由上节的电子结构分析可知,Co和Ni元素可以减弱电子杂化效应,成键的强度下降,故材料的硬度下降.
通过晶体的模量数值可以预测其本征塑性,常用的衡量指标有G/B和泊松比υ两种[20, 21]. 对于G/B的数值越小则材料的延性越好,本征塑性越高,反之,其数值越大材料的脆性越大,本征塑性越差,常用的区分材料延性和脆性的分界值为0.57. 对于纯净的Ti3Al晶体,其G/B值约为0.57,其处于延性和脆性的分界值,本征塑性较低. Co和Ni元素固溶后,Ti11Al4Co和Ti11Al4Ni晶体的G/B值分别下降为0.398和0.470,表明Co和Ni元素固溶后材料呈延性,Co和Ni元素合金化有利于提高合金体系的本征塑性. 对于Ga元素固溶后晶体的G/B值增大明显,合金体系的脆性更加明显. 泊松比υ的数值越大,材料的塑性越好. 对于纯净的Ti3Al晶体,其泊松比υ计算结果约为0.260,与实验结果吻合较好. Co和Ni元素固溶后合金体系的泊松比均增大,晶体的塑性有所提高,其中Co元素固溶后晶体的泊松比数值增大效果更加明显,其值达到0.324,表明Ti11Al4Co晶体的塑性较纯净Ti3Al晶体提高显著;而Ti11Al4Ga的泊松比数值下降较多,Ga元素固溶将降低晶体的塑性.

表4 Ti3Al-M晶体的模量(GPa)、G/B和泊松比
3.4 德拜温度
德拜温度ΘD与固体材料中共价键强度关系密切,共价键的成键越强越高,德拜温度的数值越大[21]. 利用德拜准谐模型,晶体的德拜温度可以由晶体的弹性系数进行估算.
计算所得的德拜温度列于表5,可知,Co、Ni和Ga元素后晶体的德拜温度均发生变化,其中Co和Ni元素导致晶体的德拜温度由纯净Ti3Al晶体的506 K下降为430 K和450 K,而Ga元素固溶后晶体的德拜温度上升为523 K,由德拜温度的计算结果可以推测晶体内部成键的共价性由强到弱以此为:Ti11Al4Ga、Ti3Al、Ti11Al4Ni和Ti11Al4Co,推测结果为电子结构分析结果一致. 对比弹性模量计算结果可以发现,Ti11Al4Co和Ti11Al4Ni德拜温度下降主要原因为其剪切模量和杨氏模量均较纯净Ti3Al降低.
表5 Ti3Al-M合金的德拜温度ΘD(K)
Tab. 5 The Debye temperature of (in K) of the Ti3Al-M alloys

晶体Ti3AlTi11Al4CoTi11Al4GaTi11Al4Ni德拜温度506430523450
4 结 论
采用第一性原理对合金元素Co、Ni和Ga在Do19-Ti3Al的固溶体进行研究,通过对晶体结构、电子结构和弹性常数的分析得出以下结论:
1)Ga元素倾向于置换Ti3Al晶体中Al原子,Co和Ni元素则更倾向于置换Ti原子;
2)Ga元素固溶后与Ti原子成键强度升高,而Co和Ni元素原子与Al原子之间的金属键强度下降.
3)Ga元素固溶强化效果显著,晶体的强度和硬度均提升;Co和Ni元素固溶导致合金体系的塑性显著提升,其中Co元素的效果更佳.
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
纺织科技进展(2021年8期)2021-09-01 07:39:50
毛纺科技(2020年6期)2021-01-06 03:41:48
建材发展导向(2020年15期)2020-11-26 12:55:22
工业催化(2020年5期)2020-06-23 01:59:12
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
固体火箭技术(2019年4期)2019-09-13 00:52:54
固体火箭技术(2019年3期)2019-07-31 02:53:42
四川水泥(2019年9期)2019-02-16 20:12:56
