
Mg2+掺杂锰酸锂的第一性原理研究
2019-12-06 09:31:46王云婷梁兴华吴秋满王玉江蓝凌霄
原子与分子物理学报 2019年6期
王云婷 , 梁兴华,3 , 吴秋满 , 梁 伦 , 王玉江,2 , 蓝凌霄
(1. 广西科技大学 广西汽车零部件与整车技术重点实验室,柳州 545006; 2.广西大学,南宁530005; 3. 广东省新材料研究所,广州510650)
1 引 言
锂离子电池具有比容量高、 循环寿命长、 自放电率低、 无记忆效应、 环境友好等优点[1-5]. 但是,锂离子电池的性能目前仍然无法满足日益增长的市场需求,其性能的优化往往基于正极材料技术突破,因此正极材料的研究已成为目前锂离子动力电池关注的焦点. 尖晶石型 LiMn2O4材料具有三维离子扩散通道、 放电容量大、 能量密度高、 工作电压高、 以及环境友好等优点[6],已被广泛研究并应用于锂离子电池领域. 但由于其性能仍存在局限性,如: LiMn2O4在放电过程中容易发生歧化反应,生成的Mn2+容易溶于电解液中,致使无法正常进行可逆反应、 导电性差及容量衰减严重. 因此,需要进一步改善材料的循环稳定性以满足动力电池的市场需求. 材料性能的提高有效途径是对其进行包覆或掺杂. 包覆主要是用金属氧化物对LiMn2O4材料进行表面修饰,如李秋艳等[7]通过对LiMn2O4材料进行Al2O3与SiO2双层包覆,发现包覆层在电池循环过程中缓解金属溶解问题,有效地减少界面的副反应. 另一种能有效提高锂离子电池在循环过程中的库伦效率以及能量的利用率的方法是对正极材料进行掺杂,常用的掺杂离子为三价或二价的金属离子如: Zn、 Fe、 Cr、 Co、 Al等. 实验报道[8],掺杂后的LiMn2O4材料中的一部分Ni、 Co原子被取代,当掺杂一定量Ni、 Co原子时,晶体结构的稳定性得到加强,有利于锂离子反复脱嵌锂,锂离子的扩散系数增加,最终电池的循环寿命和倍率性能得到改善.
近年来,第一性原理计算方法已成功应用于各个研究领域,并在锂电池领域受到广泛关注. 通过计算可以解释分子结构以及分子内部的信息,为阐述一些实验现象提供理论依据,从而避免了大量的盲目实验. 如郑录[9]等运用第一性原理的方法,计算了LiMn2O4及其掺杂金属离子体系的结构和电子性质,并计算了掺杂Al前后对脱锂电压平台以及结合能的影响,结果表明掺Al后会提高充放电平台以及体系结构更牢靠,由此,提高了尖晶石相LiMn2O4材料中锂离子的扩散速率、 材料的循环性能以及倍率性能. 如忻晓桂[10]等应用第一性原理计算系统地研究了LiNi0.5Mn1.5O4磁性和原子结构,详细阐述了极化子迁移机理. 本文采用第一性原理计算并分析了Mg2+掺杂LiMn2O4的电子结构. 并与实验值进行比较,解释了掺杂后对电化学性能的影响. 本文运用第一性原理计算方法对尖晶石相LiMn2O4材料掺杂前后进行研究,对其能带结构、 态密度、 分态密度、 键布居等进行计算并分析,并与实验数据进行对比,从微观结构更深层次了解LiMn2O4掺杂Mg2+后对电化学性能的影响.
2 模型构建与计算方法
2.1 模型构建
计算模型: 尖晶石型 LiMn2O4空间点群为Fd-3m,晶格常数为a=b=c=8.246 Å,α=β=γ=90°,每个晶体结构中各个原子之间均是以化学键键合的方式连接在一起. Fd-3m空间群的单晶胞内,8个锂原子占据四面体位(8a),32个氧原子占据32e位,16个锰原子占据在八面体位(16d)[11]. 利用Materials Studio软件建立LiMn2O4晶胞,掺杂时,用一个金属原子取代一个锰原子,即金属Mg掺杂后的LiMn2O4可以表示为LiMg0.125Mn1.875O4,其晶胞结构如下图所示:
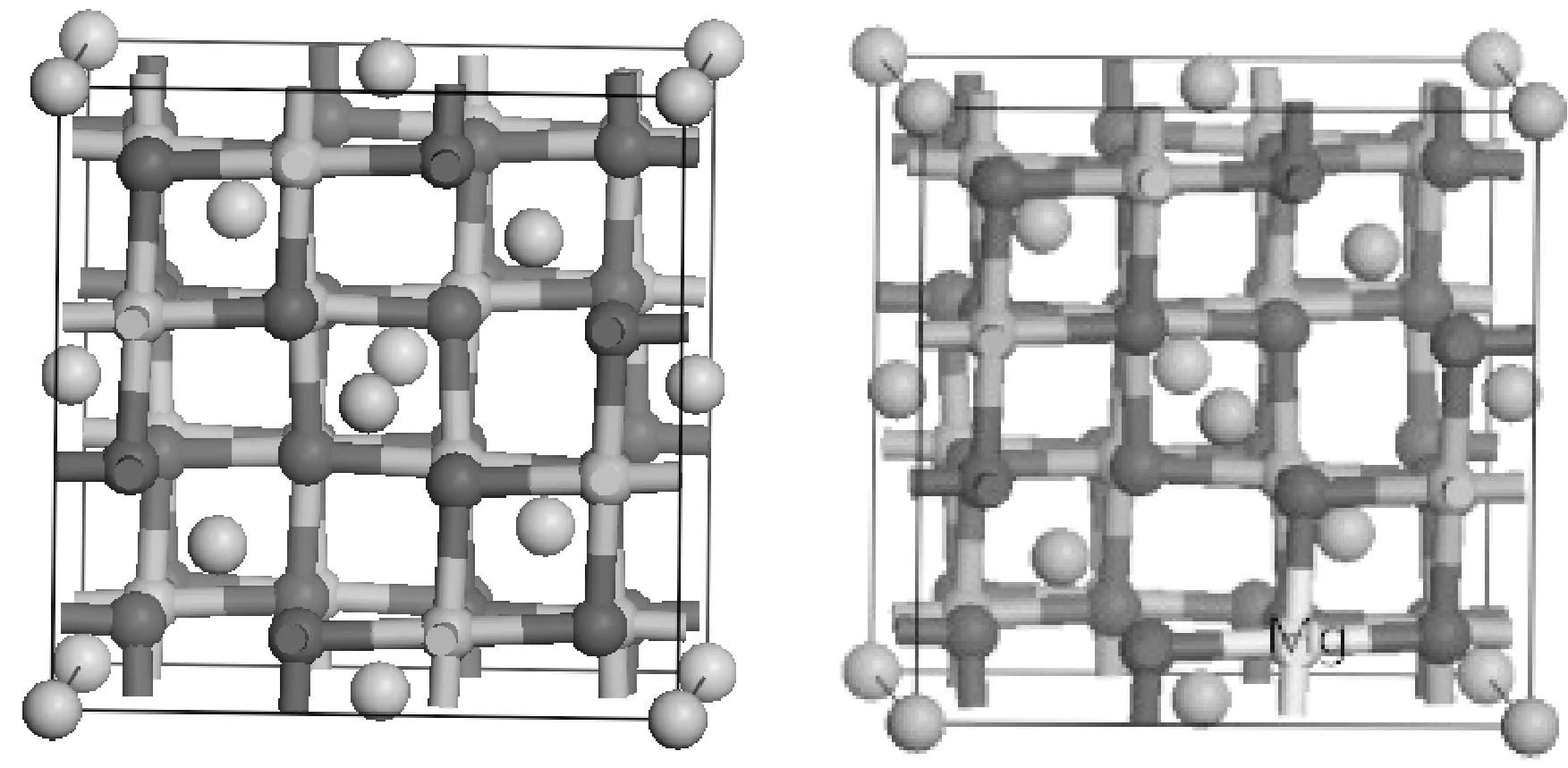
图1 LiMn2O4掺杂Mg2+前后晶胞模型Fig. 1 Cell model, before and after LiMn2O4 doping Mg2+
2.2 计算方法
本文是由Materials Studio软件中的Castep模块进行计算,该模块基于密度泛函理论[12],通过总能量平面波赝势方法[13-15],将粒子势用赝势代替,计算中电子与电子间的交换关联能采用广义梯度近似(GGA)[16]的PBE方案来描述, 电子波函数则通过平面波基矢组展开,价电子和离子实之间的相互作用势通过超软赝势 (ultrasoft pseudopoten-tial) 来描述,为了保证计算的速度和满足一定的精度,在计算过程中设置计算精度为fine,电子自洽过程中能量的收敛精度为2×10-3eV,作用在每个原子上的力不超过0.05 eV/Å,平面波展开的截断能Ecut off=340 eV,布里渊区积分计算采用5×5×5 Monkhorst-Pack型K点网格进行迭代[17,18]. 并选取Li、 0、 Mg、 Mn各原子的价电子组态为: 1s22s1、 1s22s2p4、 1s22s22p63s2、 1s22s22p63s23p63d54s2.
3 结果与讨论
3.1 几何结构优化
在软件中建立LiMn2O4晶胞后并对其进行几何结构优化,优化后晶格常数与优化前的晶格常数值如表1所示,误差仅较小,因此模型的建立是可靠的. 之后,用一个Mg原子代替一个Mn原子并对其晶胞再次进行结构优化,计算完成后将得到LiMn2O4在掺杂Mg2+前后结构优化后的晶格常数值和体积以及费米能,如表1所示,掺杂Mg2+后的晶格常数与晶胞体积都略减小,这与实验结果一致[19].
表1 LiMn2O4和LiMg0.125Mn1.875O4的晶格参数和费米能级
Table 1 Lattice parameters and Fermilevels of LiMn2O4and LiMg0.125Mn1.875O4

材料晶格参数(a/Å)体积(V/A3)费米能级/eVLiMn2O4实验值8.23557.60—LiMn2O4计算值8.24559.47-1.29LiMg0.125Mn1.875O4实验值8.21554.14—LiMg0.125Mn1.875O4计算值8.21553.38-1.02
3.2 能带结构分析
利用平面波超软赝势计算得到Mg2+掺杂LiMn2O4前后沿第一布里渊区高对称方向的能带结构图如图2(a)、 (b)所示,根据计算结果可得纯相的LiMn2O4的能带间隙为0.525 eV与文献值接近[20],比实验中得出的禁带宽度明显窄些,这主要是由于LiMn2O广义梯度近似(GGA)存在计算值普遍偏小,但能带图的整体趋势变化跟实验值相差不大,所以在同一个体系中在相同的计算环境下,使用DFT计算出来的能隙是具有可比性的. 掺杂Mg2+后的禁带宽度为0.006 eV,比纯相的LiMn2O4禁带宽度窄,较窄的带隙宽度可以有效减小电子的跃迁势垒,提高材料的导电能力.

图2 LiMn2O4与的LiMg0.125Mn1.875O4的能带结构图Fig. 2 Energy bandstructures of LiMn2O4 and LiMg0.125Mn1.875O4
3.3 态密度分析
纯相LiMn2O4与的LiMg0.125Mn1.875O4的总态密度和分态密度图如图3、 4所示,由图可知,两者的总态密度图相差不大,表明LiMg0.125Mn1.875O4仍保留一部分的LiMn2O4材料所具有的性能. 由图3可知,纯LiMn2O4的费米能级附近主要由Mn原子3d态原子和O原子2p态原子贡献,Mn-3d轨道与O-2p轨道相互重叠杂化严重,说明Mn原子和O原子的相互作用较强,而锂离子峰形尖锐说明电子局域化强,可以近似自由地嵌入脱出,这与实验事实一致. LiMg0.125Mn1.875O4体系的能带主要分布在-44.5~0.12 eV之间,总态密度主要由Mn、 O和Mg的相互作用决定的,而Li的能量跨度在-44.5~-42.7 eV之间,其锋型尖锐跨度范围小,对总态密度的贡献几乎为零,这说明Mn离子和O离子的相互作用较强,锂离子和氧离子、 锰离子的相互作用较弱,锂离子较易游离于晶体结构之中,O-2p与Mn-3d能带主要位于费米能级附近的高能量区,掺杂Mg2+后,晶体结构没有发生明显变化,而O-2p出现了新的能带,这主要是由于Mg2+起到了诱导作用使O-2p和Mn-3d的轨道成键加强,使晶体结构更加稳定,从而达到提高材料充放电循环稳定性的目的,这与实验结果一致[21].

图3 LiMn2O4的态密度图以及各元素分布图Fig. 3 Density map of LiMn2O4 and distribution of each element
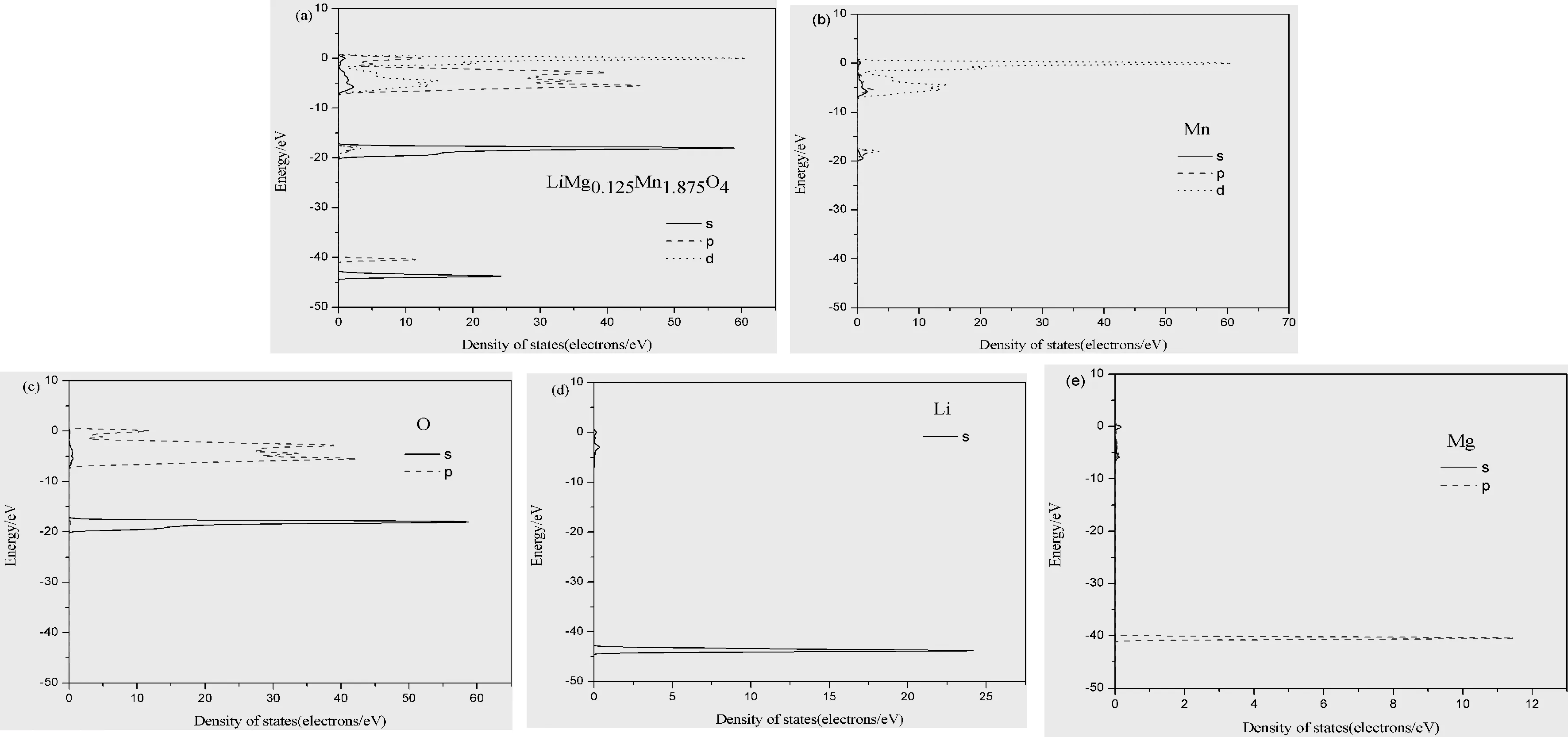
图4 LiMg0.125Mn1.875O4的态密度图以及各元素分布图Fig. 4 Density map of LiMg0.125Mn1.875O4 and distribution of each element
3.4 布局分析
Mg2+掺杂后,Mg、 O和Mn原子之间的电荷将会重新分布,Mg2+掺杂LiMn2O4前后的各电荷布居以及键长的变化情况分别如表2、 3所示,由表2数据可知计算出的原子电荷布局为+1.03 e与理论值+1e接近,说明Li+的离子化程度高,从而较多有效的Li+可以自由穿梭于正负极之间,提高了材料的导电性能. 由此解释了掺Mg2+后LiMn2O4材料电池在充放电过程中具有较大充放电比容量的宏观现象. 在表2的原子布居分布表中可以看出Mn原子的p与d轨道的电子数比掺杂前减少了,从而导致了费米能级附近的导带数增加,能缝隙减小,利于电子的能级跃迁,降低了LiMg0.125Mn1.875O4尖晶石结构在充放电过程中导致晶格坍塌发生Jahn-Teller畸变效应几率. 从表3可以知LiMg0.125Mn1.875O4中的Mn—O比LiMn2O4的短,表明锰离子与氧离子的共价性增强,形成的共价键较稳定,其相互作用形成的骨架不易坍塌. 而Li—O的键长在掺杂Mg2+后稍有变长,Li—O键能被弱化,使Li+较易脱离束缚能够自由地在晶体结构中来回脱嵌,使可逆反应得以正常进行,提高了LiMg0.125Mn1.875O4的电化学稳定性,这与相关实验结果一致[22].
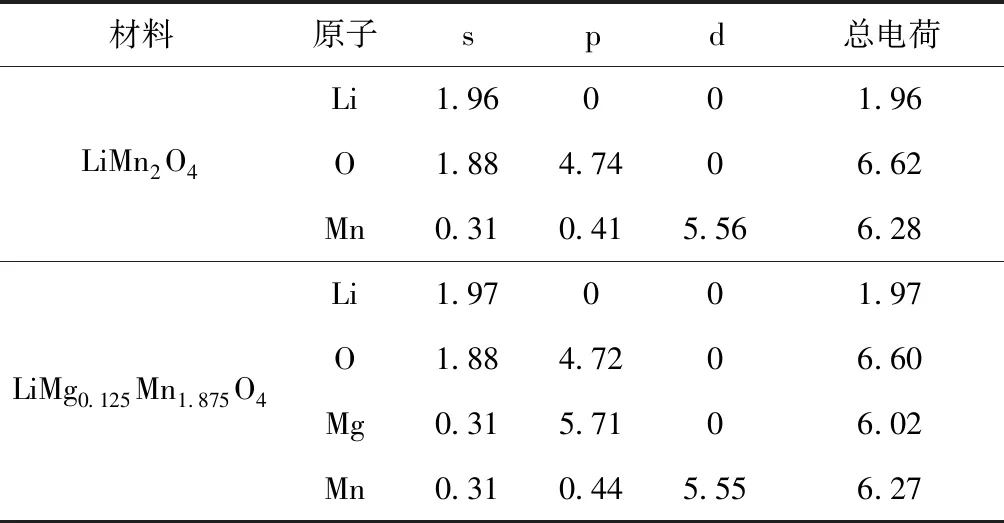
表2 原子布居分布
表3 掺杂Mg2+前后的键长变化
Table 3 Change in bond length before and after doping with Mg2+

材料M—O键长LiMn2O4Li—O1.92652Mn—O1.96866LiMg0.125Mn1.875O4Li—O1.94426Mn—O1.96671Mg—O1.95970
4 结 论
Mg2+掺杂后的LiMn2O4材料在电池的充放电循环中稳定性明显提高,从能带结构来分析主要由于掺杂Mg2+后的禁带宽度减小,利于电子的传导以及锂离子的传输. 其次O、 Mn、 Mg提供的能量高,其形成了共价键较稳定,不易断裂,使LiMg0.125Mn1.875O4尖晶石结构在充放电过程中不易发生坍塌与Jahn-Teller畸变效应,提高了循环稳定性.
猜你喜欢
高中数理化(2024年4期)2024-03-16 11:09:37
高中数理化(2022年16期)2022-09-14 13:57:04
小天使·聪聪画刊(2021年2期)2021-09-10 07:22:44
汽车零部件(2020年10期)2020-11-09 03:41:42
数学物理学报(2019年5期)2019-11-29 07:46:50
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08 03:23:36
汉语世界(The World of Chinese)(2019年6期)2019-09-10 07:22:44
数学物理学报(2017年5期)2017-11-23 07:51:09
潍坊学院学报(2016年6期)2016-04-18 13:56:55
长江大学学报(自科版)(2014年1期)2014-03-20 13:20:12
