
A2BO4型类钙钛矿材料的第一性原理研究进展
2019-12-03 03:04黄荣洲郭为民乐志文尹俊
应用化工 2019年11期
黄荣洲,郭为民,2,3,乐志文,尹俊
(1.广西科技大学 生物与化学工程学院,广西 柳州 545006;2.广西科技大学 广西糖资源绿色加工重点实验室,广西 柳州 545006;3.广西科技大学 广西高校糖资源加工重点实验室,广西 柳州 545006)
A2BO4型类钙钛矿因其具有二维层状结构和独特的物化性能,而被广泛地应用于中温固体氧化物燃料电池(IT-SOFCs)阴极材料[1-2]和功能材料等领域。目前,主要通过对该材料组成、结构进行调控,有效提高其物理性能。但是不同实验条件、组成、结构对其物理性能的影响不同,不易区分组成、结构对该材料物理性能的不同作用机制,使得通过实验难以迅速找到影响规律。第一性原理的方法提供一条途径。此外,国内外,这方面研究主要集中在这类材料的结构和磁性等性质。目前从量子层面应用第一性原理的方法解释该材料物理性质的研究报道有限。本文将综合国内外相关文献,对其的第一性原理研究进展进行综述。
1 类钙钛矿结构
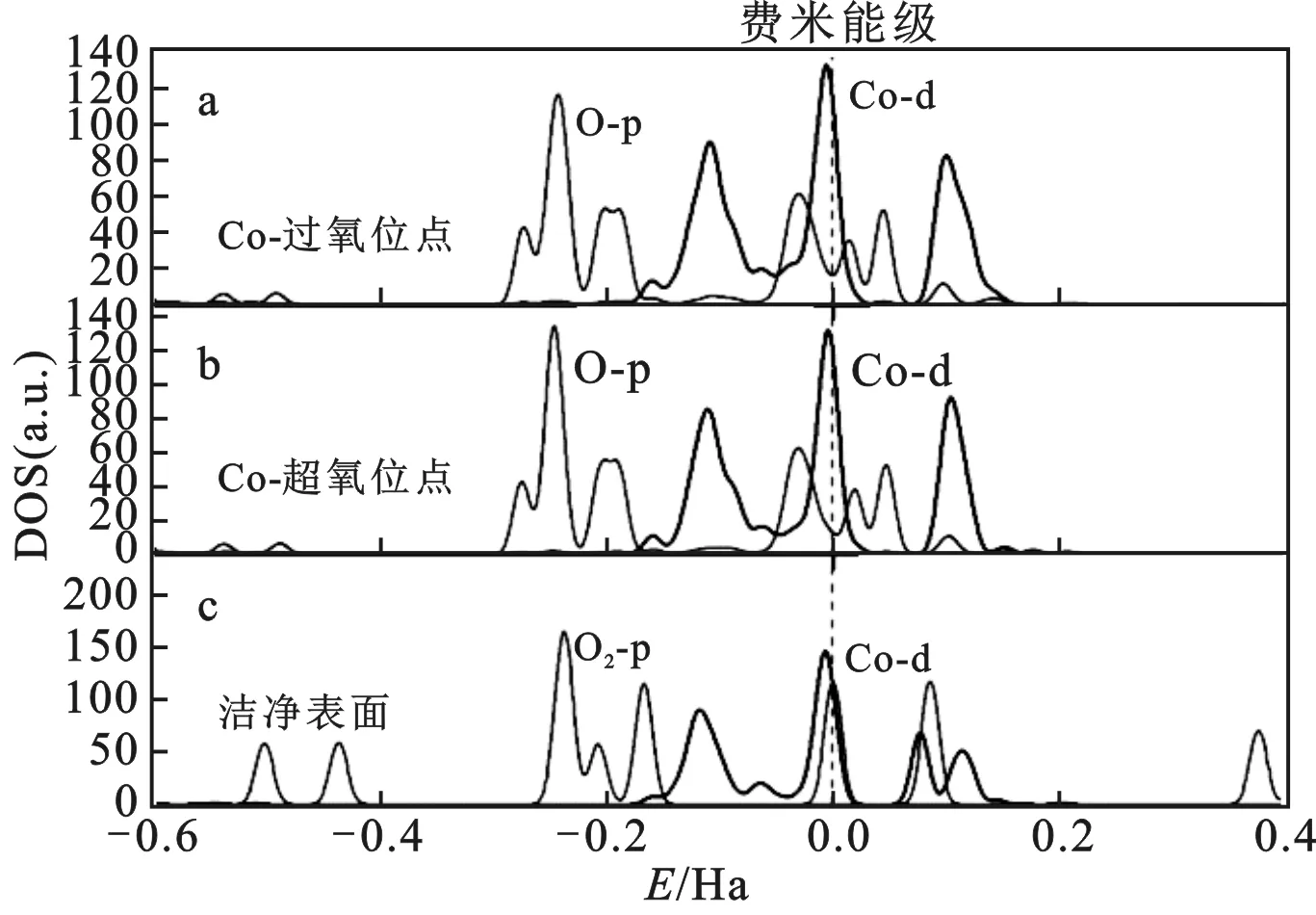
Ruddlesden-Popper(RP)系列复合氧化物是一种被广泛研究的用作IT-SOFCs阴极材料,其一般式为An+1BnO3n+1化合物[3]。该化合物中,其中A通常是大阳离子,通常是稀土金属离子或碱土金属离子;B通常是中等尺寸阳离子,通常是过渡金属离子;O为阴离子,n=1,2,3……RP相是由n个串联钙钛矿层(ABO3)n和岩盐层(AO)交替构成,岩盐层沿着晶体的c轴方向生长,其化学式可为(AO)(ABO3)n,n表示相互连接的层之间BO6八面体共用顶点数目[4]。当n=1时,即为类钙钛矿钙钛A2BO4型结构。从晶体结构看,类钙钛矿需要满足骨架的要求,A离子半径满足rA>0.090 nm,B离子半径rB>0.050 nm。结构因子(t)一般所代表形成结构的物质的稳定性。对于A2BO4型结构,满足0.85 图1 A2BO4类钙钛矿结构Fig.1 Structure of A2BO4 perovskite-likea.晶体结构;b.多面体图 层状类钙钛矿具有被三维钙钛矿结构沿某个二维平面如(010)“切割”开的结构。每两个相邻的钙钛矿层之间的共顶连接被切断,断裂处原有的A位空间变更大,层间可插入较大的阳离子或其它结构单元,调控层间距,得到新的结构。层间的次级结构通常是电子学或力学性能上的软调节层,在基本保持整个晶体结构有序不变的条件下进行调控类钙钛矿材料的性能。 第一性原理计算又称为从头计算,广义的第一性原理包含分子动力学、分子力学、蒙特卡罗、密度泛函(DFT)等方法。基于DFT可以预测A2BO4型类钙钛矿的晶格常数、态密度、电荷密度、波函数、光学性质、结构性质、布居分析分子对称性和能带结构等,其对研究类钙钛矿的特性与应用有重大的意义。目前,关于类钙钛矿材料的研究工作主要还仅限于宏观研究,而其微观研究较少。针对上述研究现状,有国内外学者分别采用了分子模拟、分子动力学模拟、计算机模拟等方法对类钙钛矿材料的微观结构与性质的内在联系进行了研究。比如对类钙钛矿的晶体结构、电子结构进行密度泛函计算,进一步从分子水平解析类钙钛矿的物理性质和化学性质。当前,关于采用第一性原理方法对类钙钛矿材料进行理论计算研究的文献报道很有限,而已见报道的文献主要针对类钙钛矿材料一些重要的性质,包括电子结构、介电性质、光学性质和迁移率等进行第一性原理方法计算。 能带结构表现出在布里渊沿高对称性的方向电子能量对k矢的依赖性,所以能带结构可用于分析材料物性[6]。其提供了对类钙钛矿材料的电子结构定性分析的工具。态密度(DOS)和偏态密度(PDOS)显示出A2BO4型类钙钛矿的电子结构的一个定性图形,提供钙钛矿电子结构重要信息的重要工具。电子的充满或不充满能带,对钙钛矿的物理性质有巨大的影响。态密度对成键的定性分析不是很强,布居是指电子在各原子轨道的分布。从布居数可以得到原子的成键情况,通过分析间布居可以获得原子与键的数据,判断成键和估计键的粒子性和共价性。 Aspera等[7]使用基于DFT的第一性原理计算研究了Pr2NiO4+δ(δ=0和0.031)的结构和电子性质,分析了属于空间群I4/mmm的高温四方(HTT)相和属于空间群Bmab的低温正交(LTO)相的两种结构,发现这两种结构的差异主要是由于在Bmab结构中NiO6八面体发生了显著的扭曲而导致的。而这种扭曲是引起准金属性的HTT结构转变为金属性的LTO结构的原因。通过分析HTT结构中Pr2NiO4的局域态密度(LDOS),观察到该结构是自旋极化的且具有准金属性质。同时,也发现其大部分自旋态具有金属性质,这是由Pr、Ni和O原子轨道穿越费米能级而引起的;而费米能级附近的小能隙中的少数自旋态则显示出半导体性质;尽管在HTT结构中观察到自旋极化特征,但是在LTO结构中所有的自旋态都显示出金属性质;LTO结构与HTT的相似,这些DOS主要来自Pr和Ni原子,它们将沿着ab平面表现出各向异性的电子电导。此外,Yao等[8]对于处于基态的SrGdCoO4进行了第一性原理方法计算,结果表明,该化合物自旋极化高,费米能级处的主要DOS大部分来自于Co原子的贡献,且该化合物的自旋极化也来自Co原子的影响。Yao等[9]基于DFT对Sr1.5La0.5CoO4进行了能带结构计算,结果表明,在费米能级处形成了一个明显的自旋向下的能带,这说明该化合物具有高自旋极化特性。Wang等[10]基于DFT对Sr2-xRExCoO4(RE=Gd和Y,x=0.1~0.5)高压相的能带结构进行了计算,研究表明未掺杂和掺杂化合物的电子结构均表现出高自旋极化。Yao等[11]利用第一原理对Sr2-xNdxCoO4(x=0.5,0.75,1,1.25)计算,结果表明Nd掺杂的Sr2CoO4化合物显示出高自旋极化。通过综合分析上述文献报道的计算结果,可以利用第一性原理方法计算从微观方面预测或解析A2BO4型类钙钛矿的性质。其中,在电子结构方面,可从能带结构、DOS、PDOS和LDOS等深入分析该物质的机理,比如上述报道根据物质的高自旋极化性质可分析出来源于哪些原子的贡献,以及可比较掺杂与未掺杂物质的性质,为掺杂离子取代改善材料的性能提供有力的理论支撑。 Zhou等[12]使用DFT并结合周期平板模型对在LaSrCoO4中氧空位的形成和强氧吸附动力学机制进行了研究,DOS分析结果表明,Co和O之间形成了强杂化,分子吸附相应的Co—O键长减小。这表明,由于LaSrCoO4的氧空位形成能和迁移能量低,使得其有望成为固体氧化物燃料电池的阴极材料。图2为游离和吸附O2的DOS。其中,图2a和2b分别对应过氧、超氧类物质吸附后的DOS。由图可知,与游离O2相比较,吸附O2带向较低能量方向移动;Co原子的d轨道都参与Co—O的键合,Co—d和O—p轨道的DOS峰形成重叠揭示了两原子轨道之间存在强烈杂化,同时也表明Co原子的d轨道对于Co-O2-S/P构型的键合起重要作用。Zhou等[13]建立了高温四方相La2NiO4块体和表面模型,通过第一性原理计算研究其电子结构,发现DOS穿越费米能级,具有典型的金属特性,价带由Ni 3d和O 2p态构成,而导带主要由La 5d态构成。Forslund等[14]使用DFT对La0.5Sr1.5Ni1-xFexO4±δ进行了计算,结果表明,穿越费米能级的eg(Ni)、eg(Fe)和2p(O)带之间形成的交叉间隙杂化可增强Fe—O—Ni键桥间的电荷转移相互作用,以及可增加带宽有利于电极吸附物的电子转移。Yun等[15]利用DTF对平面波超软赝势原理计算,研究了Sr2TiO4的电子结构,结果表明,角共享的TiO6八面体主导了Sr2TiO4的主要电子性质,在ab平面上Ti—O(1)键的共价性强于沿c轴的Ti—O(2)键的共价性。上述针对A2BO4型类钙钛矿材料的第一性原理方法计算研究,通过对材料的DOS和布居等进行了分析,结果认为:通常,原子与原子之间的布居数小,其键长,说明共价性弱、离子迁移率强和结构相对不稳定;相反,布居值大与键长短,说明其共价性强与结构较稳定。此外,通过DOS和PDOS的分析能为分析布居分析提供帮助。基于上述分析,A2BO4型材料的第一性原理方法计算,可为此类材料在固体氧化物燃料电池阴极材料、催化和光学等方面的应用研究提供有益的参考数据。 图2 LaSrCoO4(010)表面吸附O2的Co位点的态密度[12]Fig.2 Density of states of Co site on which O2 is adsorbed with the Co-super and Co-per configurations for the LaSrCoO4(010) surface 介电常数是介电材料通过极化屏蔽外部电场的能力的一个重要参数。在电介质常数中存在两个贡献:它与电子密度(光学)的重组和振动离子的极化有关,其中涉及离子运动。以往在光伏器件中使用的典型半导体实验表明,对于宏观介电常数(电子学贡献)来说,大于10的值可以很好地将激子解离成自由载流子。 Fengie等[16]利用DFT研究了Sr2TiO4的介电性质,得到了关于响应的各向异性的信息,这种各向异性是解释陶瓷和薄膜样品的介电响应的现有实验数据的关键,其研究结果确定Ti—O键在晶格对响应的贡献中起主导作用。Parker等[17]用DFT研究了单斜晶系β-Ba2TiO4,得出未表现出强的极性晶格畸变,这导致其平均静态介电常数的相当低的值。Zhou等[18]利用DFT计算了LaSrAlO4的介电性质,计算结果表明LaSrAlO4的频率依赖性复介电常数。在介电函数的实部ε1(ω)中存在两个峰值,它们的位置分别在5.1 eV和19.0 eV和0~30 eV的范围内。在虚部中,ε2(ω)中存在3个主峰,它们与带内跃迁相关,其中带内跃迁被忽略,因为仅在金属材料中考虑了带内跃迁。从上述例子可知,介电常数是A2BO4型材料的重要参数,可用于分析该类材料的介电性质,对于评价介电材料的性质具有重要的理论意义。 Yun等[15]利用DFT平面波超软赝势的第一性原理计算,研究了掺入对Sr2TiO4的光学性质,Sr2In0.125Ti0.875O4体系的价带(VBS)由O 2p和4d态组成,O 2p和4d态的混合使顶部VBS显著地向高能量移动,从而产生可见光吸收。可见光的吸附对于Sr2TiO4作为光催化剂的应用具有重要的现实意义。Yun等[19]利用DFT对平面波超软赝势的第一性原理计算,研究了La掺杂对Sr2TiO4的光学性质,该物质的DOS向低能量转移,光学带隙变宽。Sr1.875La0.125TiO4透明性高,在可见光范围内的透光率约为90%,比Sr0.875La0.125TiO3的(85%)高。这是由于费米能级中杂质的PDOS导致的宽带隙、小跃迁几率和弱吸收导致了薄膜的光学透明性。Yun等[20]基于DFT对平面波超软赝势进行第一原理计算,研究了Nb掺杂对Sr2TiO4的光学性质,计算结果表明,当x=0.125,由于电子掺杂,费米能级转变为SrNbTiO的导带,系统表现为N型简并半导体特征;Sr2TiO4在其主晶轴上表现出光学各向异性,而c轴显示了获得宽透明区最合适的晶体生长方向;在Sr2Nb0.125Ti0.875O4的可见光范围内,光学透光率高于90%。Yun等[21]通过DFT计算研究了Sc掺杂Sr2TiO4的光催化性能,与未掺杂的Sr2TiO4相比,Sr2Sc0.125Ti0.875O4的带隙窄,约0.25 eV,这导致光谱吸收带边红移,见图3。特别指出,掺杂后Sr2Sc0.125Ti0.875O4的导带和价带的色散增强,这对于提高半导体材料光催化性能是非常重要的。此外,在可见光区域出现了新的弱吸收,这将有利于扩大Sr2Sc0.125Ti0.875O4的光响应范围。通过上述分析,可知采取合适的掺杂方法,对材料的某个离子进行取代,可以调控其的化学组分和维持晶体结构不变,通过改变材料能带结构来提高其性能。第一性原理计算方法,可以从原子和电子水平上解析掺杂离子对其的影响和相关机理。对于改进材料的性能,特别是在光催化性能方面能提供理论指导。 图3 Sr2TiO4(a)和Sr2Sc0.125Ti0.875O4(b)的DOS与PDOS[21]Fig.3 DOS and PDOS of Sr2TiO4(a) and Sr2Sc0.125Ti0.875O4(b) 迁移率是评价材料的电化学性能的一个重要指标之一。大量研究表明,材料迁移率越高,则材料的电化学性能越优异。Petrov等[22]在NaLnTiO4(Ln=La,Nd)层状钙钛矿型化合物中,利用交流阻抗谱研究了钠离子的迁移率,并用分子动力学方法模拟了钠离子的迁移率,在523~923 K的温度范围内研究了NaLaTiO4和NaNdTiO4晶体,研究了钠原子扩散系数与温度的关系,结果表明因为NaLaTiO4中化学键的离子性更强,所有钠离子在NaLaTiO4中的迁移率比NaNdTiO4中的高。Naumovich等[23]使用静态晶格和分子动力学(MD)方法的理论计算模拟方法,研究了阴离子的迁移路径、相关能量参数和过渡金属阳离子掺杂剂如何影响La2Ni(M)O4+δ(M=Fe,Co,Cu)固溶体中氧离子输运的机制,结果表明,特别是掺入+3氧化态的掺杂剂会导致更高的离子电荷载流子浓度从而影响阴离子的扩散率,这主要是由在层状晶格中占据正常位置的阴离子和间隙氧的协同机制决定的。然而,Fe、Co、Cu掺杂剂趋向于降低A2BO4型结构的岩盐和钙钛矿层中的阴离子迁移率。所以,在对A2BO4材料进行晶体结构调控时,可对所掺杂离子的扩散、迁移路径和离子输运等性质进行模拟计算和分析,能为实验中此类材料的结构调控与优化选择掺合适的掺杂离子类型提供理论指导,从而提高材料性能。 上述研究报道表明,第一性原理方法计算在A2BO4型类钙钛矿材料研究领域发挥着重要的作用,也为该领域积累了许多崭新的研究成果。第一性原理计算方法在研究A2BO4型类钙钛矿材料的各种性质时既能精确获得丰富的参考数据,又能拓展人们在微观方面对该类材料的性质更进一步地认识。但是上述的研究尚处于纯粹的理论研究阶段,需要理论和实践相结合,从而更好地为该类材料的研究提供参考数据和理论支撑。 当前,在A2BO4型类钙钛材料实验研究中侧重电化学性能研究较多,但如何从晶体结构和电子结构不同结构层面去认清类钙钛矿材料电化学性能调控的内在规律,揭示各种因素对电学性质的影响机制,这是一个需要攻关的难点。所以,很有必要从电子原子、分子和电子水平上更进一步地深入研究A2BO4型类钙钛矿型的晶体结构、电子结构、磁性质、铁电性质、光学性质和热学性质等,特别是对此类材料的晶体结构和电子结构方面细节的深入认识,能有力地揭示材料的性质。可以预见,A2BO4型类钙钛矿的第一性原理方法研究仍然将是该领域的研究热点之一,可为此类材料各种性能的应用研究提供理论依据。

2 类钙钛矿材料的第一性原理研究
2.1 A2BO4型类钙钛矿材料的电子结构
2.2 A2BO4型类钙钛矿材料的介电性质
2.3 A2BO4型类钙钛矿材料的光学性质和光催化性能


2.4 A2BO4型类钙钛矿材料的迁移率
3 展望
猜你喜欢
陶瓷学报(2021年2期)2021-07-21
中学生数理化(高中版.高二数学)(2021年5期)2021-07-21
中等数学(2020年6期)2020-09-21
原子与分子物理学报(2020年5期)2020-03-17
中等数学(2019年6期)2019-08-30
中学生数理化·七年级数学人教版(2018年4期)2018-06-28
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
燕山大学学报(2015年4期)2015-12-25
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年3期)2015-11-24
