
不同高分子材料及有机溶剂体系对阿司匹林缓释微球的影响
2019-11-28 05:01周雪肖潮达贺智勇姜丰吴林菁肖婷沈祥春陶玲
贵州医科大学学报 2019年11期
周雪, 肖潮达, 贺智勇, 姜丰, 吴林菁, 肖婷, 沈祥春, 陶玲
(贵州医科大学 贵州省天然药物资源高效利用工程中心 贵州省高等学校天然药物药理与成药性评价特色重点实验室 贵州医科大学-贵阳市联合重点实验室 天然药物资源优效利用重点实验室 药学院, 贵州 贵阳 550025)
阿司匹林(aspirin,Asp)已被广泛应用了100余年。近年来,小剂量Asp在抑制动脉粥样硬化[1-2]、预防和治疗心肌纤维化[3-4]、抑制肺纤维化[5-6]、抗肿瘤[7-8]及心血管疾病等[9-11]方面发挥了新的作用,但目前Asp的上市剂型均需要长期多次给药,血药浓度波动大、胃肠道副作用大、病人的顺应性差,限制了其临床疗效的发挥。微球(microspheres,MS)作为一种新型递送系统,其给药途径广泛,具有延缓或控制药物释放、增加制剂稳定性、提高药物生物利用度、降低毒副作用、提高疗效等优势[12-14],且合成可生物降解聚合物作为新型MS载体材料,由于其良好的生物相容性和生物可降解性,已被广泛认可和应用[15-16]。由于药物的物理化学性质不同,使用不同的高分子材料可以显著改变药物的载药量和包封率[17-18]。本研究以包封率、载药量为考察指标,对聚乳酸-羟基乙酸共聚物(PLGA)、聚乳酸(PLA)、聚己内酯(PCL)和聚乙二醇-聚(乳酸-羟基乙酸)嵌段共聚物(PEG-PLGA)4种高分子材料对Asp 缓释MS进行考察,另就2种不同有机溶剂体系对MS制备的影响进行了更全面的药剂学性能研究,为制备不同要求的MS提供参考。
1 材料与方法
1.1 材料与仪器
S-3400N型扫描电子显微镜(日本Hitachi公司),XDS-1B型倒置显微镜(重庆麦克光电仪器有限公司),Bettersize2000型激光粒度分布仪(丹东百特仪器有限公司),STA-449C型差示扫描量热仪(德国Netzsch公司),D/Max-2200型全自动X射线衍射仪(日本理学公司),Nicolet 6700型傅里叶变换红外分析仪(美国Thermo Fisher Scientific公司),JNM-ECS400型核磁共振谱仪(日本JEOL公司);Asp原料药[阿拉丁试剂(上海)有限公司,批号D1316056,纯度99%],Asp对照品(中国药品生物制品检定所,批号100113-201104,纯度99.9%),PLGA、PLA、PEG-PLGA及PCL(济南岱罡生物工程有限公司,相对分子质量均为30 kDa),乙酸乙酯、丙酮、二氯甲烷等均为分析纯。
1.2 方法
1.2.1Asp缓释MS的制备 (1)高分子载体材料的考察:根据药物的理化性质和聚合物材料的性质,选择4种不同类型的聚合物PLGA,PLA,PEG-PLGA,PCL,同时选取对MS制备工艺影响较显著的3个因素[投药比、O/W体积比及油相比例(DCM ∶ACE)]进行考察,以载药量及包封率为主要考察指标,优选高分子载体材料。(2)Asp-PEG-PLGA-MS的制备[19]:根据前期实验结果得到有机溶剂体系混合最佳比例,采用O/W型乳化溶剂挥发法制备Asp-PEG-PLGA-MSs,称取适量Asp及PEG-PLGA溶解于1 mL混合有机溶剂体系中(DCM ∶ACE为3 ∶1 或 DCM ∶EA为1 ∶1),涡旋混匀,作为油相。在1 000 r/min搅拌条件下将油相注入1% 聚乙烯醇(PVA)Asp饱和溶液中,冰浴条件搅拌,缓慢加入Asp水饱和溶液120 mL,20 min后调至室温800 r/min搅拌4 h,静置沉淀,去除上清液,抽滤,蒸馏水洗涤3次,室温干燥,得到不同溶剂体系制备的MS。(3)MS载药量及包封率的测定:称取适量MS样品,加入适量二氯甲烷,涡旋、离心、重复上述操作3次,合并上清液,二氯甲烷定容至25 mL;采用紫外分光光度法在波长276 nm处检测吸光度,计算浓度,并照下式计算Asp缓释MS的载药量及包封率。载药量=(MS中含药量/最终MS实际重量)×100%,包封率=(MS中药物含量/投入药物量)×100%。
1.2.2MS的表征 (1)MS外观形态观察及粒径测定:在倒置显微镜下,直接取少量MS观察外观形态;同时,将MS用离子溅射仪进行表面喷金处理,采用扫描电子显微镜(SEM)观察,对MS的外观和表面形态进行评价;室温条件下,取Asp-PEG-PLGA-MSs混悬液适量,用激光粒度分析仪测定MS的粒径及其分布。(2)差示扫描热分析(DSC):称取Asp、PEG-PLGA、空白MS、Asp与各组空白MS的物理混合物,以及实验制得的Asp-PEG-PLGA-MSs各10 mg,将上述样品各两种分别进行DSC分析,测试条件为0~180 ℃,升温速率为10 ℃/min,氮气氛20 mL/min。(3)X射线衍射分析(XRD):各称取Asp、PEG-PLGA、空白MS、Asp与各组空白MS的物理混合物,以及实验制得的Asp-PEG-PLGA-MSs适量,将上述样品各2组分别进行XRD分析,进一步观察Asp在载体材料PEG-PLGA中的分散状态,测试条件为Cu靶,管电压/电流为40 kV/30 mA,扫描速度4°/min,扫描范围为0~80°。(4)核磁共振分析(NMR):分别称取2组Asp、PEG-PLGA、空白MS、Asp与各组空白MS的物理混合物,以及实验制得的Asp-PEG-PLGA-MSs各8.0 mg,溶于0.5 mL氘代氯仿中,在室温下以1H-NMR 分析在制备过程前后Asp的核磁谱图及研究制备过程对Asp的影响。(5)红外光谱分析(FT-IR): MS中Asp与PEG-PLGA的相互作用通过傅立叶变换(FT-IR)进一步分析,分别称取上述两组的Asp、PEG-PLGA、空白MS及实验制得的Asp-PEG-PLGA-MSs,加入适量干燥KBr粉末,充分混合均匀,再转入模具中,分布均匀,抽真空下压成透明薄片;装入压片夹进行 FT-IR扫描,在400~4 000 cm-1波长范围内进行扫描。
1.2.3AspMS体外释放度的考察 (1)体外累计释放度的测定:采用动态透析法进行Asp-PEG-PLGA-MSs体外累计释放度的测定。精密称取各组MS 40 mg置入透析袋中,置于具塞锥形瓶中,加入PBS缓冲液50 mL,(37.0±0.5)℃恒温水浴摇床以100 r/min的速度振摇,在一定时间点上分别取出PBS 5 mL,同时补加等量同温的PBS缓冲液,随后测定吸光度,计算累积释放百分率,绘制释放曲线。(2)载药MS体外释药模型拟合:由于168 h后两组载药MS累计释放百分率均接近100%,故采取零级动力学模型、一级动力学模型、Higuchi模型、Riger-Peppas模型,对优选处方制备的MS 168 h内体外释药曲线进行拟合。
2 结果
2.1 Asp缓释MS的制备
2.1.1高分子载体材料的考察 如表1所示,PEG-PLGA制备的MS载药量和包封率均最高。
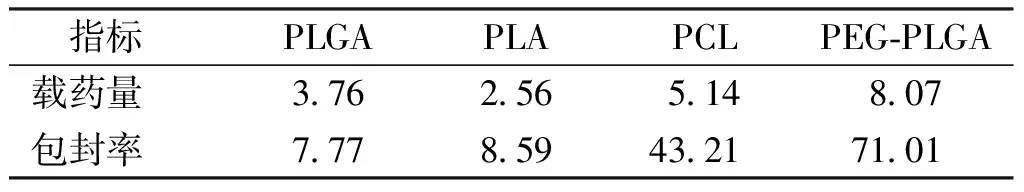
表1 不同聚合物对AspMS的载药量和包封率的影响(%)Tab.1 The drug loading and encapsulation efficiency of aspirin microspheres with various p polymers
2.1.2不同溶剂体系制备Asp-PEG-PLGA-MS比较 如表2结果所示,以DCM ∶EA为有机相制备所得Asp-PEG-PLGA-MSs平均载药量、包封率及收率均明显高于DCM ∶ACE组MS;反复实验证实,当溶剂体系为DCM ∶EA,Asp用量为10 mg时,包封率均>120%,这可能与不同有机溶剂体系导致高分子材料对外水相中游离药物的吸附能力不同有关,将进一步通过核磁共振分析来验证。此外,在常规搅拌4 h除去有机溶剂后,MS在洗涤和收集过程中不黏附;然而,24 h后产生明显黏附现象,可能是内部有机溶剂提取不完全所致;因此,本研究将MS静置过夜,低速搅拌2 h后再进行收集,黏附现象明显改善,且不影响载药量。

表2 不同溶剂体系制备MS的载药量、包封率及收率比较Tab.2 Different binary solvent systems for optimal microsphere preparation
2.2 MS的表征
2.2.1MS外观形态观察及粒径测定 光学显微镜观察结果表明,2种溶剂体系制备的Asp-PEG-PLGA-MSs均成球圆整,分布均匀。扫描电子显微镜下可见(图1),MS成球性良好,表面较光滑,略有小凹陷,可能为制备过程中有机溶剂挥发所致;室温条件下,取Asp-PEG-PLGA-MSs混悬液适量,用激光粒度分析仪测定MS的粒径及其分布,结果表明,以DCM ∶ACE及DCM ∶EA制备的ASA-PEG-PLGA-MSs粒径分别为(100.9±2.52) μm及(142.4±3.25) μm。
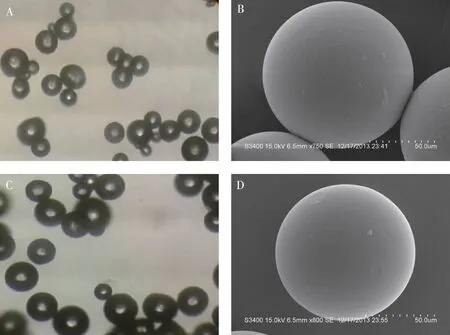
注:A、B为DCM ∶ACE, C、D为DCM ∶EA;A、C为光学显微镜下(40×),B、D为扫描电子显微镜下(800×)。图1 光学显微镜与扫描电子显微镜下Asp-PEG-PLGA-MSs形状Fig.1 Light micrographs and scanning electron microscopy photographs of Asp-PEG-PLGA-MSs
2.2.2DSC结果 如图2所示,2种溶剂组DSC结果无明显差异,Asp与各组空白MS的物理混合物均有2个明显的放热峰(32~37 ℃为PEG-PLGA,142~145 ℃为Asp),然而Asp-PEG-PLGA-MSs只有一个明显的放热峰(32~37 ℃为PEG-PLGA),另一个所代表Asp的放热峰消失了,说明制备所得MS并不是2种物质的简单物理混合。

图2 Asp-PEG-PLGA-MSs的DSC结果Fig.2 The DSC results of Asp-PEG-PLGA-MSs
2.2.3XRD结果 如图3所示,Asp、Asp与各组空白MS的物理混合物的特征图谱在0~30°均有很强的Asp结晶衍射峰;而两组不同溶剂体系制得Asp-PEG-PLGA-MSs的图谱中无明显的Asp结晶衍射尖锋,说明Asp-PEG-PLGA-MSs并不是简单的物理混合,可能Asp制成MS后其晶型发生了变化,药物可能以无定形状态分散于载体材料中。

注:A为DCM ∶ACE, B为DCM ∶EA。图3 Asp-PEG-PLGA-MSs的XRD分析结果Fig.3 The XRD results of Asp-PEG-PLGA-MSs
2.2.4NMR结果 如图4所示,Asp的δ 7.14、7.35、7.62和8.12分别为苯环的次甲基(—CH—)质子峰,δ 2.34为甲基(—CH3)质子峰;PEG-PLGA的δ 5.20和1.65 分别为PLA的次甲基(—CH—)和甲基(—CH3)质子峰,δ 4.81为PGA的亚甲基 (—CH2—)质子峰,δ 3.63为PEG的亚甲基(—CH2—)质子峰;同时,空白MS、Asp与PEG-PLGA物理混合物均在相应位置有质子峰,而Asp-PEG-PLGA-MSs在δ 6.90、6.95、7.48与7.90也有质子峰的出现,这表明Asp可能向高场发生了化学位移,其可能是Asp的晶型改变所致。
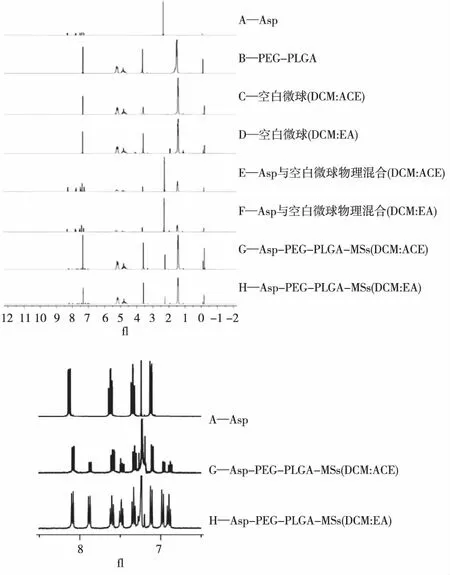
图4 Asp-PEG-PLGA-MSs的1H-NMR结果Fig.4 The 1H-NMR results of Asp-PEG-PLGA-MSs
2.2.5FT-IR结果 如图5所示,在Asp原料药的IR图谱中,Asp明显的特征峰均存在:δ= 1 696 cm为环上COOH羰基伸缩,δ=1 758 cm-1为接苯环CH3CO中的酯羰基伸缩,δ=1 610、1 570、1 480 cm分别为苯环的C骨架伸缩;高分子材料PEG-PLGA的IR谱中,δ=1 760 cm-1处有强吸收峰,为酯羰基C=O的伸缩振动吸收峰,在δ=3 502 cm-1附近宽吸收峰为两端的OH吸收峰,δ=2 881 cm为C-H的伸缩振动吸收峰;2类空白MS以及Asp-PEG-PLGA-MSs的IR谱中,特征峰未发生明显改变;说明Asp制备成MS后,药物与PEG-PLGA结构中的主要基团未发生改变。

图5 Asp-PEG-PLGA-MSs的FT-IR结果Fig.5 The FT-IR results of Asp-PEG-PLGA-MSs
2.3 AspMS体外释放度考察
2.3.1体外累计释放度 如图6所示,分别以一定比例的DCM ∶ACE与DCM ∶EA为溶剂的最佳工艺制备所得MS,在体外释放实验中释放MS所含80%药物所需时间分别不低于96 h与72 h,完全释放分别需要不低于120 h与168 h,而Asp原料药在同一条件下,24 h释放基本完全,说明MS有较好的缓释作用。

图6 不同溶剂体系制备Asp-PEG-PLGA-MSs的体外释药曲线Fig.6 In vitro release profile of Asp-PEG-PLGA-MSs prepared with different solvent systems
2.3.2载药MS体外释药模型拟合结果 如表3所示,Riger-Peppas 模型对2组MS释药曲线拟合的相关系数均最大,故2组Asp-PEG-PLGA-MSs的体外释药比较符合Riger-Peppas释药模型。DCM ∶ACE组,lnQ=0.455 6 lnt+2.225 9、r2=0.982 2;DCM:EA组,lnQ=0.466 5 lnt+2.248 7、r2=0.980 8,即Asp从MS中释放方式均为溶蚀型。

表3 Asp-PEG-PLGA-MSs 体外释放模型拟合Tab.3 The fitting result of Asp-PEG-PLGA-MSs in vitro release
3 讨论
本研究在对PLGA、PLA、PCL 及PEG-PLGA几种高分子材料优选中,以PEG-PLGA作为MS载体时,载药量和包封率明显高于其他高分子材料,这可能与PEG-PLGA的两亲性,以及Asp具有的微小溶解性能(水中溶解度为3 g/L)有关。
不同有机溶剂具有不同的物理性质,如黏度、表面张力、水溶性及蒸汽压等,都直接影响MS的粒径大小、载药量、包封率、稳定性及体外释放。有机溶剂溶解度越高,向水中扩散速度越快,而乙酸乙酯的溶解度介于二氯甲烷和丙酮之间;蒸汽压越高,脱除速率越快,导致聚合物的沉积与固化速率加快[20],乙酸乙酯的蒸汽压低于丙酮和二氯甲烷。因此,乙酸乙酯弥补了丙酮被快速萃取,将药物一并带出的不足,进而提高了载药量和包封率。另外,采用DCM ∶EA制备Asp-PEG-PLGA-MSs,投药量在10 mg时,包封率均超过100%,原因可能是在上述溶剂特性的影响下,当乳滴进入PVA饱和Asp溶液后,MS固化过程中,外水相中的游离Asp被吸附于MS表面所导致。
本研究的DSC和XRD分析结果表明,所制得MS并不是药物与载体材料的简单物理混合,而是药物可能以无定形状态分散在载体材料中。此外,本研究还采用NMR与IR考察Asp与PEG-PLGA在不同溶剂中的化学结合,结果发现Asp与PEG-PLGA在主要功能基团上未发生结合,但是IR图谱中表明,2组空白MS和高分子材料PEG-PLGA在一些非功能基团上发生了一定改变。
体外累计释放度的测定结果显示,2组MS体外初始释放时,释放药物速度均较高,可能是由于MS表面吸附的Asp在释放初期迅速解吸附导致。此外,由于MS体外初始释放时药物浓度较高,具有较大的势能,更加促进药物的扩散[21]。24 h后,随着表面药物的完全释放,药物MS的释放随之受到载体材料降解速度的限制,使MS以较为恒定的速度缓慢释放药物。另外,2组MS的体外释药在48~96 h内略有不同,药物从DCM ∶EA组MS释放较DCM ∶ACE组稍多,这可能与其载药量和粒径大小相关:DCM ∶EA组MS的载药量几乎是DCM:ACE组MS载药量的两倍,DCM ∶EA组MS的粒径超过DCM ∶ACE组MS的40%,有明显更大的比表面积。
综上,综合上述各项性能指标结果可知,不同高分子材料和有机溶剂体系共同影响载药MS的粒径、形态、载药包封及释放行为。这为制备不同要求的AspMS提供了参考价值与依据。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13
婚姻与家庭·婚姻情感版(2021年6期)2021-06-01
中国兽医杂志(2018年9期)2018-12-29
中成药(2018年9期)2018-10-09
中成药(2017年5期)2017-06-13
医学信息(2016年36期)2017-02-23
癌变·畸变·突变(2016年4期)2016-08-22
海峡科技与产业(2016年3期)2016-05-17
小说月刊(2015年6期)2015-12-16
中国卫生标准管理(2015年32期)2015-07-18
