
围脂滴蛋白基因CRISPR/Cas9载体的活性分析
2019-11-21 11:09许祥董维鹏张少华冯晨毅刘田福燕炯
生物技术通报 2019年11期
许祥 董维鹏 张少华 冯晨毅 刘田福 燕炯
(1.山西医科大学公共卫生学院,太原 030001;2.山西医科大学动物实验中心,太原 030001)
CRISPR/Cas9技术是近年来新出现的一种基因修饰技术,得益于其操作简单、高效率和低成本的优势在基因编辑领域被广泛应用[1-2]。通过改良的CRISPR技术,已经可以实现基因敲除、基因沉默、基因敲入以及各种目的的基因编辑。CRISPR/Cas9系统元件sgRNA和Cas9蛋白,可以通过不同载体作用于目的基因,无论是质粒、慢病毒或者直接转染sgRNA与Cas9蛋白的混合物,都可以很方便的实现基因编辑[3]。由于sgRNA的切割活性受到众多因素影响[4-6],且CRISPR/Cas9系统允许错配的出现,所以获取高效的sgRNA并验证其切割活性是实现靶向编辑的关键。
脂滴结构蛋白Perilipin蛋白高表达于白色脂肪组织中[7-8],主要功能是保护脂滴中的脂质免受脂肪酶的作用,帮助脂质在脂滴中聚集[9]。围脂滴蛋白(Perilipin1,PLIN1)是一种存在于脂滴表面可被磷酸化的蛋白,其表达量和肥胖成显著正相关,并且在高血压等代谢性疾病的发生发展过程中发挥重要作用[9-10]。前期研究发现,在基础状态下,PLIN1对脂滴发挥屏障作用,可以有效降低甘油三酯的分解,当其含量降低或者发生磷酸化时,就可以加快甘油三酯的分解,且其基因的表达受到PPARγ的调控,是脂质代谢的一个重要环节,但其具体调节机制尚不清楚。本课题基于CRISPR/Cas9技术,设计了3条理论切割效率最高的sgRNA序列,并对其进行体外切割活性验证和细胞转染基因敲除效率验证。得到了可以高效率切割PLIN1基因的sgRNA序列,并初步探究了PLIN1对脂解的影响,旨为后续PLIN1基因敲除动物模型的建立奠定基础。
1 材料与方法
1.1 材料
3T3-L1 前脂肪细胞系、FBS(胎牛血清)、DMEN/F12细胞培养基、Western blot相关试剂(武汉BOSTER生物工程公司),大肠杆菌DH5a感受态细胞、琼脂糖、DNA Marker、细胞基因组DNA提取试剂盒、PCR 实验相关试剂(北京天根生化科技有限公司),限制性内切酶、T4 PNK、Quick Ligase(美国NEB公司),sgRNA 体外转录试剂盒、Cas9 体外酶切试剂盒(苏州泓迅生物科技股份有限公司),琼脂糖凝胶DNA回收试剂盒(北京聚合美生物科技有限公司),CRISPR 基因敲除表达载体(PX459)(美国Addgene公司),氨苄青霉素(合肥博美生物科技有限公司),蛋白Maker(北京聚合美生物科技有限公司),Perilipin 1抗体(英国Abcam公司)。
1.2 方法
1.2.1 sgRNA的设计与合成 根据NCBI中公布的小鼠PLIN1基因序列,应用在线工具(http://crispr.mit.edu)设计可以识别PLIN1基因2号外显子的gRNA序列。根据脱靶位点、基因数和发生错配的可能性大小进行分析,给出所有序列。选择3条特异性较好的gRNA序列(表1)作为实验序列。
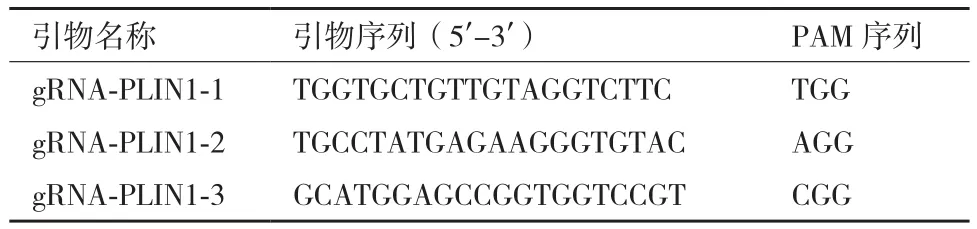
表1 guideRNA序列
将 T7(TAATACGACTCACTATAGGG) 启 动子添加到设计好的gRNA序列的5'端,保守序列(GTTTTAGAGCTAGAAATAG)添加到3'端作为体外转录上游引物,由上海生工公司合成。
1.2.2 Cas9/gRNA表达载体的构建及鉴定 选择CRISPR基因敲除质粒PX459(图1)作为载体,在gRNA scafflod位置加入目的sgRNA序列。氨苄青霉素抗性基因用于质粒转化与扩增过程的筛选,嘌呤霉素抗性基因用于转染过程的筛选。在3对PLIN1gRNA的5'端添加黏性末端后,将得到的PLIN1 sgRNA上下游引物配置成100 mmol/L,各取1mL,加入10×T4 ligation Buffer 1 mL,T4 PNK 0.5 mL,定容到10 mL。37℃孵育30 min进行激酶处理,95℃孵育5 min,缓慢降温至25℃。用限制性内切酶BbsI在PX459质粒2个BBS酶切位点(第 245、267位置)切割质粒,使其线性化。用1%的琼脂糖凝胶电泳进行检测,并用琼脂糖凝胶DNA回收试剂盒进行回收线性化质粒载体。将sgRNA退火产物与回收的线性化载体用T4连接酶进行连接,连接体系于16℃过夜。构建好的质粒载体转化至DH5a菌群中,2 d后挑取单克隆送至北京微旋基因有限公司进行检测。构建好的3个质粒载体分别命名为sgRNAPLIN1-1、sgRNA-PLIN1-2、sgRNA-PLIN1-3,并以未构建的PX459质粒作为阴性对照载体。
1.2.3 sgRNA序列的体外切割活性验证
1.2.3.1 PCR体外扩增转录模板 利用3条PLIN1gRNA特异性体外转录上游模板和gRNA通用下游引物SG-R Primer,将人工合成的通用gRNA模板的DNA片段作为模板,通过PCR特异性扩增PLIN1的体外转录模板。扩增产物使用2%的琼脂糖凝胶电泳进行检测,并使用DNA纯化试剂盒进行纯化。
1.2.3.2 sgRNA体外转录 将扩增好的DNA转录模板按表2顺序依次加入,充分混匀,37℃孵育4 h(可适当延长孵育时间)。转录完成后取少量反应液稀释20倍,用2%的琼脂糖凝胶电泳检测产物的大小及完整性。使用RNA纯化试剂盒进行纯化,通过微孔板分光光度计测定转录产物的的浓度和纯度。
1.2.3.3 Cas9体外酶切 构建Cas体外酶切体系,将得到的sgRNA与Cas9蛋白以及PLIN1基因片段(PLIN1-F:5'-GCAGAGGTAGACAGCCCAAG-3';PLIN1-R :5'-TCCTGCCACTGGTCCTACTC-3') 混合孵育。设立阴性对照组(未加sgRNA),sgRNAPLIN1-1实验组,sgRNA- PLIN1-2实验组,sgRNAPLIN1-3实验组,37℃孵育30-120 min。孵育完成后使用DNA纯化试剂盒进行纯化。纯化结果进行琼脂糖凝胶电泳观察切割效率。
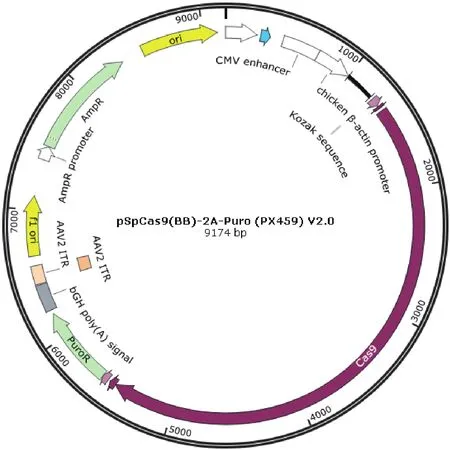
图1 CRISPR/Cas9质粒表达载体
1.2.4 CRISPR/Cas9质粒载体细胞内活性鉴定
1.2.4.1 T3-L1细胞的培养 用含10%FBS的DMEM高糖培养基培养3T3-L1前脂肪细胞:放置于37℃、5%CO2的恒温培养箱中,2 d更换培养液1次。传代时用胰酶消化细胞,并转移细胞至10 mL无菌离心管中,800 r/min离心5 min。将细胞沉淀平均分到两个新的含有5 mL完全培养基的培养瓶中继续培养。

表2 体外转录体系
1.2.4.2 电转染 在电转染前一天,细胞传代,使细胞在转染前融合度达到 80%。收集细胞沉淀至 EP管中。加入 500 mL 1640 培养基,充分吹打均匀。取10 mL细胞悬浮液,显微镜下观察并计数。调整细胞浓度至 7×106/mL 左右,加入7 mg/mL 的质粒,吹打均匀。在0.2 mm电转杯中加入含有质粒的细胞悬液 100 mL,按照预定条件设置参数(波型:指数波,电压:160 V,电阻:500F),进行电穿孔。将电转杯放入细胞恒温培养箱中10 min,使质粒充分进入细胞。取出电击杯,在预热的细胞培养基(不含双抗)中接种细胞悬液,摇晃均匀,放入培养箱中正常培养。待细胞贴壁后,更换培养基继续培养。
1.2.4.3 嘌呤霉素筛选 细胞电转染24 h后,用已确定的嘌呤霉素筛选培养基培养细胞。2 d更换嘌呤霉素培养基一次,继续培养4 d,随时观察细胞生长状态。转移少量筛选后的细胞到一个新的细胞培养瓶中,用完全培养基继续培养7 d。
1.2.4.4 诱导分化 将细胞接种于6孔板中,待细胞汇合至80%左右时,按本课题组前期优化过的经典“鸡尾酒”方法,诱导分化3T3-L1前脂肪细胞成为成熟的脂肪细胞[8]。首先用诱导分化培养基Ⅰ培养细胞2 d,再用诱导分化培养基Ⅱ继续培养2 d,随后更换为完全培养基继续培养4 d,每2 d换液1次。
1.2.4.5 实时荧光定量PCR检测细胞中PLIN1 mRNA的表达 当细胞回合至80%左右的时候,PBS冲洗,收集到EP管中。按照试剂说明书的要求,提取总RNA,逆转录为cDNA,然后进行实时荧光定量PCR扩增,以β-actin为内参,用2-ΔΔCt值表示目的基因的相对表达量。PLIN1基因cDNA RT-PCR引物,上游:5'-GAGAGGAGACAGACGACGAGGAG-3',下游:5'-GGTCACTGCG GAGATGGTGTTC-3',交上海生工公司合成。
1.2.4.6 Western blot检测细胞中PLIN1蛋白的表达量 收集各组细胞沉淀至EP管中,加入200 μL细胞总蛋白裂解液,冰上裂解10 min,4℃、12000 r/min离心15 min,上层液体即为待测总蛋白样品。采用BCA法测定各组待测样品的总蛋白浓度并调平。PAGE电泳,电泳参数:浓缩胶80 V恒压,分离胶120 V恒压;湿式转膜,参数:220 mA恒定电流,转膜持续2 h。取出NC膜,浸入封闭液中封闭2 h。蛋白一抗4℃过夜孵育。次日取出条带,孵育二抗,37℃摇床2 h。显影仪曝光目的条带,挑选目的条带清晰、背景浅的图片。用ImageJ软件对目的蛋白条带进行分析并测定吸光度值,以β-actin进行校正。
1.2.5 PLIN1基因敲除对3T3-L1细胞脂解的影响 细胞干预完成后,收集6孔板中的细胞至EP管。每个EP管中加入900 μL PBS缓冲液,用枪头吹打细胞沉淀至细胞悬浮。在冰水浴条件下进行超声破碎细胞。制备好的匀浆液直接按照甘油三酯(TG)试剂盒检测说明书测定TG含量,每组以细胞总蛋白浓度对测定值进行校正。制备好的匀浆液70℃金属浴10 min,灭活脂肪水解酶,严格按照甘油测定试剂盒说明测定样本甘油含量。根据预先绘制的标准曲线计算各样本的甘油含量,并以每mg蛋白浓度对测定值进行校正。
2 结果
2.1 CRISPR/Cas9质粒干扰载体构建及鉴定
构建的CRISPR/Cas9质粒载体进行靶位点测序,测序结果与预期目标一致(图3),sgRNA序列成功连接到到质粒载体上,表明质粒载体构建成功。
2.2 sgRNA体外切割活性鉴定
通过PCR获得带有T7 Promoter的sgRNA体外转录模板,经琼脂糖凝胶电泳检测分子量在120 bp左右,与预期结果一致。对转录模板进行纯化回收,浓度为44±3.1 ng/mL,A260/A280比值为1.89±0.04,证明体外转录模板构建成功。用sgRNA体外转录模板进行体外扩增,3条sgRNA转录体外转录产物均在100-200 bp之间(图3),与预期结果一致。对转录产物进行纯化回收,浓度全部大于5000 ng/mL,A260/A280比值为2.03±0.06,证明体外转录sgRNA成功。
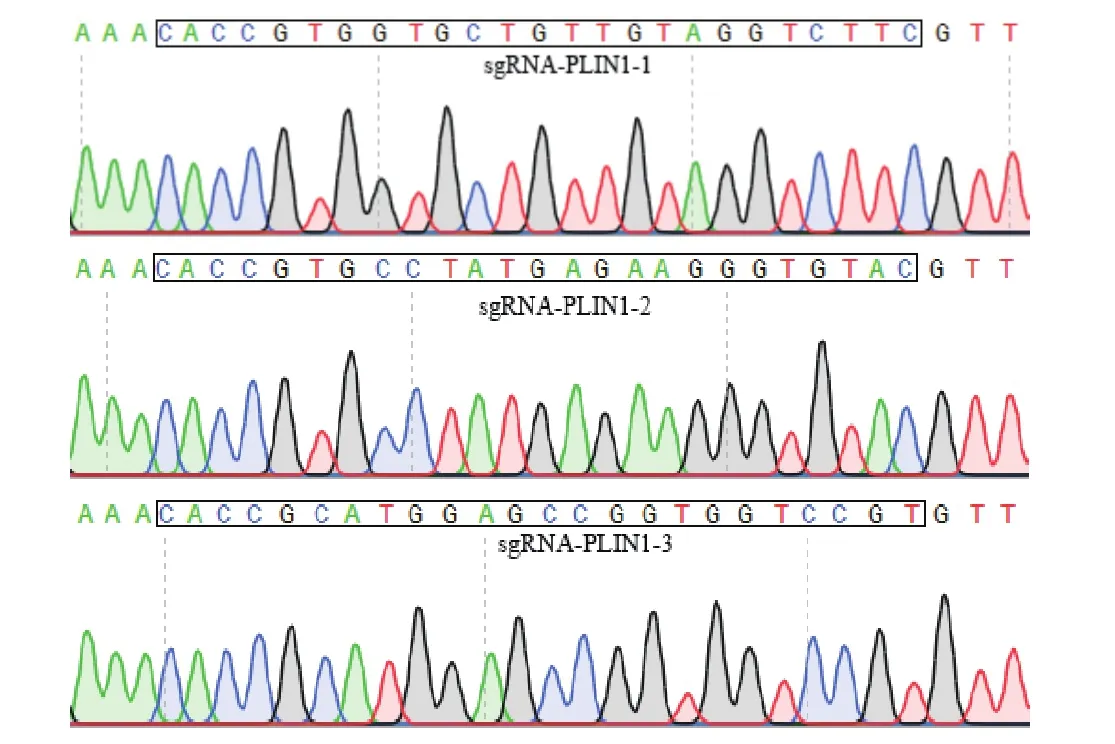
图2 CRISPR/Cas9质粒载体sgRNA测序结果
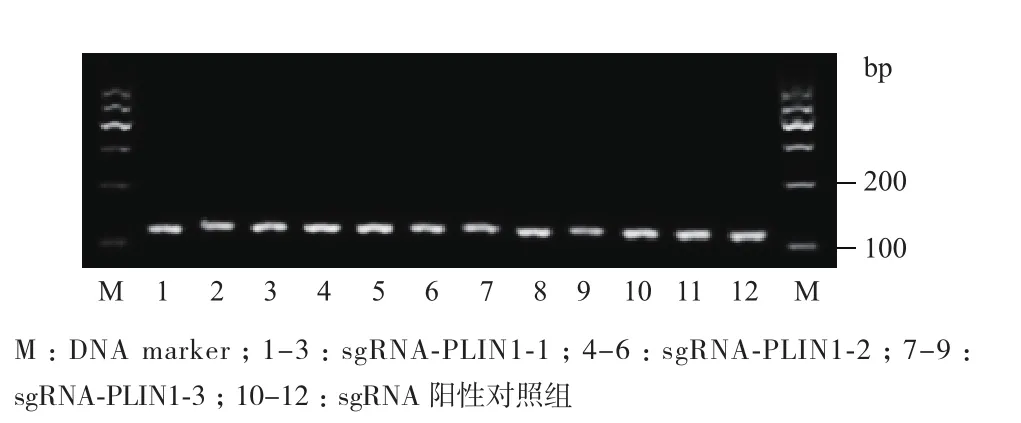
图3 sgRNA体外转录产物电泳
以扩增好的PLIN1基因为底物,分别用3条sgRNA进行构建体外酶切体系,反应1 h后,底物被切割为2个条带(图4)。利用灰度值计算切割强度,得sgRNA-PLIN1-1的切割效率为61.8%±9.0%,sgRNA-PLIN1-2的 切 割 效 率 为64.1%±9.6%,sgRNA-PLIN1-3的切割效率为34.1%±7.2%。计算公式为,其中 fcut=切开的条带强度之和/全部条带强度之和。3组sgRNA均可成功切开底物PLIN1基因,其中sgRNA-PLIN1-3组的切割效率明显低于sgRNAPLIN1-1组和sgRNA-PLIN1-2组,差异有统计学意义(图5)。
2.3 3T3-L1前脂肪的诱导分化
显微镜下观察,3T3-L1前脂肪细胞呈梭形,内部无明显脂滴,诱导分化可以使细胞趋向圆形,并逐渐出现脂滴,油红O染色后脂滴呈现“戒环样”(图6)。电转染构建好的sgRNA-PLIN1-1、sgRNAPLIN1-2、sgRNA-PLIN1-3质粒载体和阴性对照质粒,12 h后贴壁,存活率均为80%左右,无显著差异。嘌呤霉素杀死曲线确定嘌呤霉素 4 d 杀死细胞的最低浓度是 4 mg/mL;嘌呤霉素筛选细胞后,存活率均为30%左右,无显著差异。
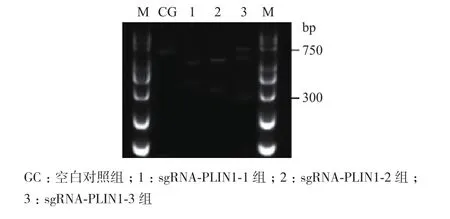
图4 体外酶切琼脂糖凝胶电泳
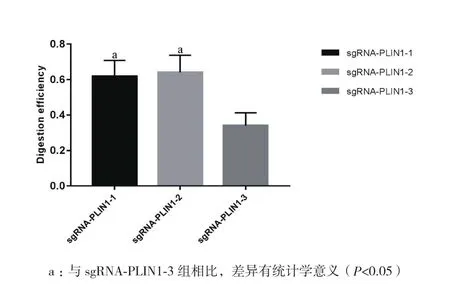
图5 体外切割效率比较
2.4 3T3-L1脂肪细胞中PLIN1 mRNA的表达
在诱导分化的第4天,检测sgRNA-PLIN1-1组、sgRNA-PLIN1-2组、sgRNA-PLIN1-3组、阴性对照组的PLIN1的mRNA表达量。如图7所示,各阳性组中PLIN1mRNA表达量显著降低,差异有统计学意义。sgRNA-PLIN1-1组和sgRNA-PLIN1-2组的mRNA表达量较sgRNA-PLIN1-3组降低,差异有统计学意义。
2.5 3T3-L1脂肪细胞中PLIN1蛋白的表达
在诱导分化的第4天,检测sgRNA-PLIN1-1组、sgRNA-PLIN1-2组、sgRNA-PLIN1-3组、阴性对照组和空白对照组的PLIN1的蛋白表达量。如图7所 示,sgRNA-PLIN1-1组、sgRNA-PLIN1-2组 和sgRNA-PLIN1-3组的PLIN1蛋白几乎不表达,差异无统计学意义。3组sgRNA阳性组与阴性对照组相比蛋白表达均有明显降低,差异有统计学意义。3组sgRNA阳性组与空白对照组相比蛋白表达均有明显降低,差异有统计学意义(图8)。
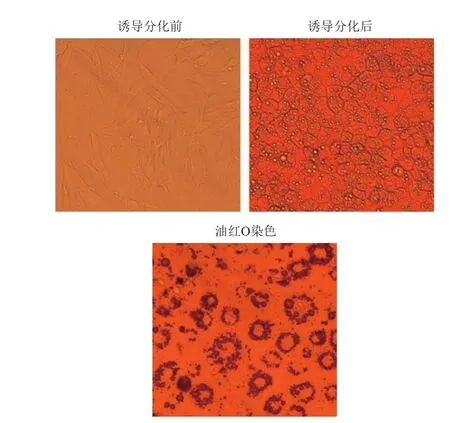
图6 3T3-L1细胞的诱导分化
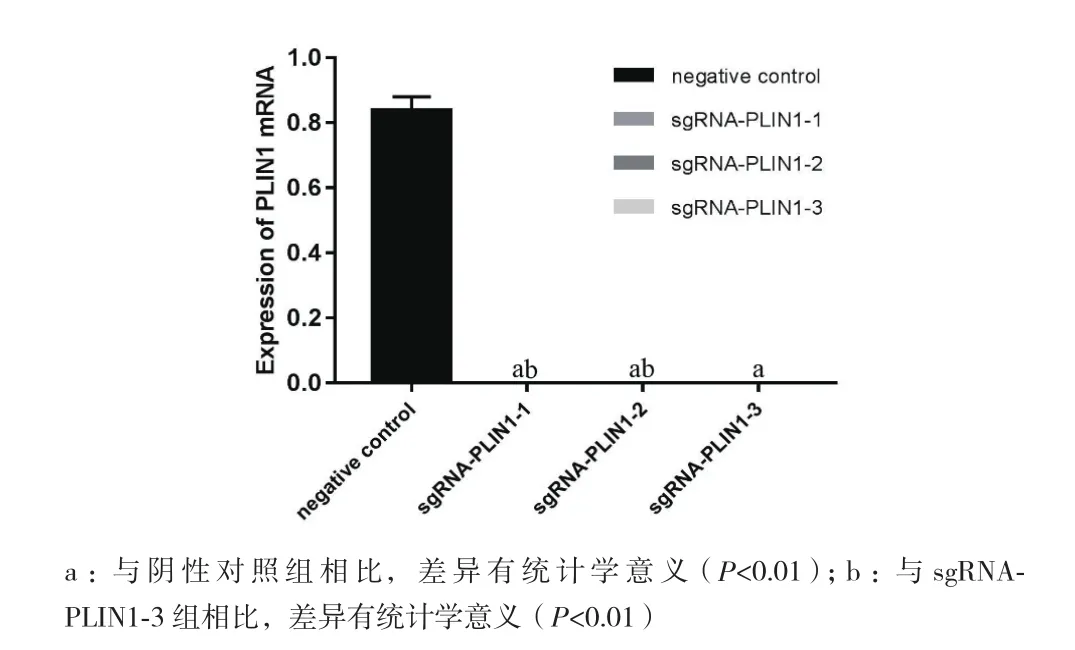
图7 PLIN1 mRNA相对表达量
2.6 PLIN1基因敲除对脂解的影响
各组细胞诱导分化第8 天油红O染色,随机选择50个细胞,用Image pro plus软件测量各个细胞中脂滴直径大小。与对照组相比,阳性敲除组中小脂滴数量增加,中脂滴、大脂滴数量减少,差异有统计学意义(图9)。测定各组细胞中的甘油三酯以及甘油含量,结果(表3)显示与对照组相比PLIN1基因敲除组中甘油三酯含量较低,甘油含量较高,差异有统计学意义。

图8 PLIN1蛋白表达相对量
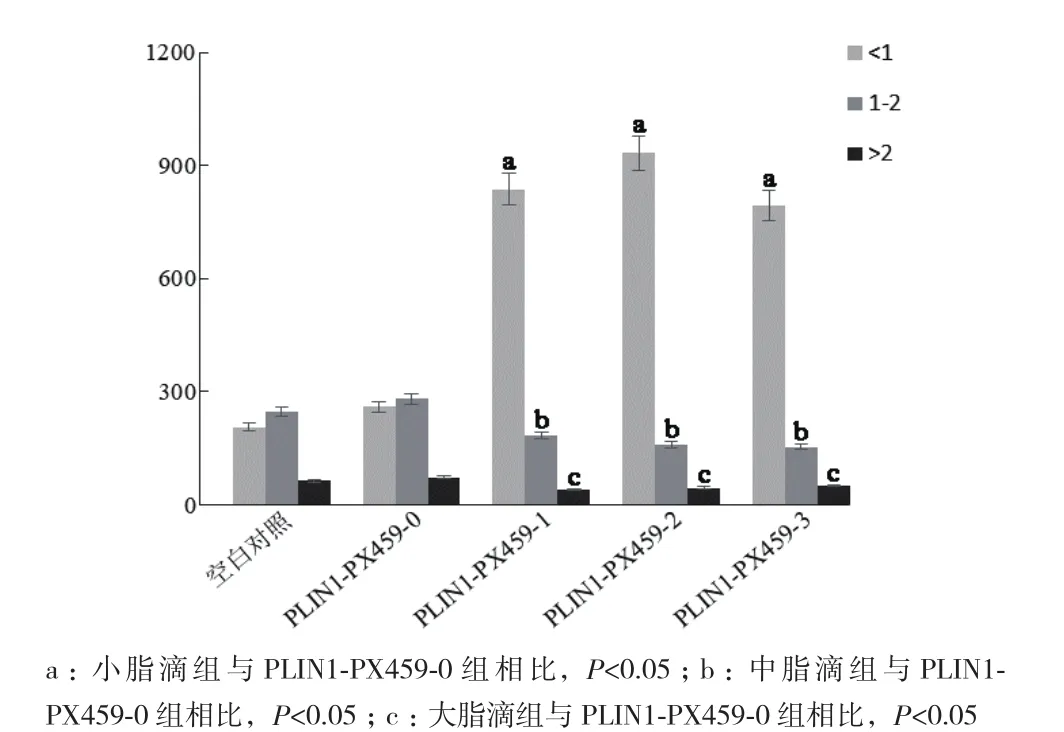
图9 脂肪细胞中脂滴大小数量

表3 脂肪细胞中TG与甘油含量
3 讨论
脂质代谢紊乱会引发一系列慢性疾病,如肥胖症、2型糖尿病、心血管疾病、脂肪肝等,严重影响了人们的生命健康,降低了人们的生活质量[11]。研究者普遍认为,脂滴作为能量的贮存器,是一个复杂的、活动旺盛的、动态变化的多功能细胞器,脂滴内含有甘油三酯酶,它与其他细胞器(线粒体)相互作用,在脂质代谢与储存、膜转运等过程中发挥作用[12]。围脂滴蛋白(Perilipin1,PLIN1)是脂滴表面蛋白的重要一员,完全定位在脂滴表面,在脂肪生成与脂滴成长过程中发挥重要作用[13]。本实验可以明显观察到PLIN1基因的敲除会抑制小脂滴的聚集,促进甘油三酯的水解。
研究表明切割越靠前的外显子导致移码突变的概率越大,本次实验前部针对PLIN1基因的2号外显子设计sgRNA序列。体外活性切割实验相较于细胞实验具有高效、快速、低成本的优势。在实验中,3条sgRNA序列皆可以靶向切割PLIN1基因的2号外显子,但sgRNA-PLIN1-3组的效率明显低于另外两组。且通过观察3条sgRNA切割位置发现,它们在目的基因上相距较近,且PAM序列都有较强的引导性,但切割效率却有差异,说明影响其活性的因素很多。所以设计多条sgRNA并验证其体外切割活性是有意义的。为进一步验证体外活性的准确性,将3条sgRNA通过电转染转入3T3-L1前脂肪细胞中,验证其在细胞内的切割活性。细胞内切割以后会引入双链断裂,发生同源重组。通过嘌呤霉素筛选转染成功的细胞,3条sgRNA阳性组的PLIN1 mRNA和蛋白表达量均有显著降低。RT-PCR实验结果显示,gRNA-PLIN1-1组和sgRNA-PLIN1-2组相较与sgRNA-PLIN1-3组有更高的活性。这证明体外切割活性好的sgRNA序列在细胞内仍保持较高的活性。两组实验可以相互预测,这与吴曦等[14]的实验结论一致。mRNA水平的差异并未导致蛋白表达水平有明显差异,但是可以明显观察到蛋白表达量水平的降低,可能原因是sgRNA的靶点基因组,mRNA水平的检测敏感度高于蛋白水平。
4 结论
本研究证明了PLIN1基因可以促进脂解,设计了3条PLIN1基因的sgRNA,并构建其基因敲除载体。通过体内和体外活性实验,证明了体外切割活性和体内切割活性有一致性,验证了3条sgRNA的切割活性,得到了2条切割效率更高的sgRNA,为后续的基因敲除动物模型的构建奠定了基础。
猜你喜欢
中国预防兽医学报(2022年6期)2022-12-29
家畜生态学报(2022年2期)2022-02-28
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
食品科学(2020年4期)2020-03-11
天津医科大学学报(2019年6期)2019-08-13
现代检验医学杂志(2016年2期)2016-11-14
听力学及言语疾病杂志(2015年5期)2015-12-24
