
转基因玉米MON89034、MON810、MIR162双重数字PCR定量方法的建立
2019-11-15 09:33梁文杨镇州李妍罗超闻艳丽刘刚
中国测试 2019年6期
关键词:生物技术
梁文 杨镇州 李妍 罗超 闻艳丽 刘刚

摘要:针对进出口贸易中涉及较多的转基因玉米MON89034、MON810、MIR162品系,研究一套双重数字PCR(dPCR)定量检测方法,包括引物探针的序列设计和浓度,DNA模板浓度,PCR反应过程的时间、温度等。该方法的定量限为0.1%,转基因定量检测范围覆盖0.1%~100%,线性系数为0.999,精密度优于10%。该检测方法中,每个微反应体系都含有两套引物探针,分别用FAM和VIC荧光通道进行检测,能实现内外源基因的同时检测,避免同一样品因取样不一致造成的定量差异。该方法可以同时应用到市面上最常用微滴dPCR平台和3D芯片dPCR平台,且两种方法定量结果一致性好。
关键词:生物技术;数字PCR;双重数字PCR;转基因玉米
中图分类号:0503 文献标志码:A 文章编号:1674-5124(2019)06-0070-07
收稿日期:2019-01-02;收到修改稿日期:2019-03-18
基金项目:国家科技重大专项项目(2018ZX0801112B);国家自然科学面上基金项目(21775104);上海市质量技术监督局科研项目(2017-03)
作者简介:梁文(1986-),女,四川德阳市人,工程师,硕士,主要从事转基因检测方法研究、核酸定量方法研究。
通信作者:刘刚(1982-),男,山东青岛市人,教授级高工,博士,主要从事核酸检测方法研究。
0 引言
随着生物技术产业的迅速发展,全球转基因作物的研发速度和面积持续增加,转基因的食品安全问题也成为世界关注的热点[1]。控制转基因的含量,实施转基因食品标识制度是各国加强转基因管理的重要举措。转基因检测方法分核酸检测和蛋白检测两类。核酸在生物细胞中含量相对稳定,在产品加工中也相对不容易被破坏,而且核酸检测方法灵敏度高、操作简便,因此成为转基因检测的主流方法[2]。常用的核酸检测手段包括等温扩增、普通PCR、多重PCR、实时荧光定量PCR(real-timefluoresence quantitative,qPCR)、数字PCR(dPCR)等方法[3]。目前常用的qPCR法可以筛选转基因特异DNA片段,但大多数的qPCR在转基因检测方法的应用还停留在定性阶段[4]。
dPCR作为一种新的PCR技术在各检测领域发展迅猛,尤其在转基因定量检测方面有独特优势。dPCR的技术特点是,不需要建立工作曲线,避免了标准品配制过程及标准品与待测样品的天然差异导致的偏差[5]。dPCR采用终点荧光检测,微小反应单元的独立扩增不易受到转基因样品DNA提取过程中抑制物的影响[6]。dPCR定量准确度高,对于痕量转基因DNA样品检测重复性好[7]。在dPCR检测体系中设计双重荧光检测,分别用于标记外源基因和内源基因,可以实现内外源基因的双重检测,进一步避免加样误差的同时降低了检测成本,提高了检测效率。
本文在转基因玉米品系MON89034,MON810,MIR162定量检测体系中实现了内源基因和外源基因的同时检测,通过一个反应直接得出外源基因和内源基因的拷贝数,进而给出外源基因和内源基因的拷贝数浓度的比,即转基因的含量[8]。目前,市面上的dPCR分为微滴PCR和芯片PCR两大类。两者在独立的PCR反应微小单元形成方式,加热介质方面各有不同。本文首先在芯片dPCR平台上建立了实验方法,考察其特异性、定量限和线性范围、精密度。最后将该方法应用到微滴dPCR中,考察了两种平台的结果一致性。
1 实验部分
1.1 试剂和仪器
植物基因组DNA提取试剂盒(天根DP305,北京);引物及探针(佰力格,上海);3D数字PCR系统(QuantStudioTM3D,ABI公司);微滴数字PCR(QX200,伯乐公司);核酸定量仪(Nanodrop2000,thermo公司)。
转基因标准物质:转基因玉米种子粉末MON810(ERM-BF413gk),转基因玉米种子粉末MON89034(AOCS 0906-E),转基因玉米种子粉末MIR162(AOCS1208-A),种子粉末均为F1代杂合子。
1.2 转基因DNA的抽提
用TIANGEN公司的植物基因组DNA提取试剂盒进行玉米基因组DNA的抽提。在抽提过程中需要用到酚氯仿,以有效去除玉米材料中的多糖和多酚成分。抽提结束后用核酸定量仪定量DNA的浓度,要求其A260/A280在1.8~2.0之間。
1.3 DNA模板浓度的控制
dPCR法的模板DNA浓度受到泊松分布原理的限制,即大量微反应单元(10000以上)中,DNA模板的分布应具备随机性。由于目前市售dPCR的微反应单元数量都至少高于10000个,在保证定量结果的重复性优于25%的条件下,要满足定量限达到0.1%,则模板DNA浓度应保证上样体系中总DNA模板数(内源基因拷贝数)在4700~73551拷贝范围内[9]。从而避免低转基因含量样本出现转基因片段漏取样的情况,也避免DNA拷贝数过高,阳性过载使数据精确度下降的情况。
1.4 反应体系和反应条件
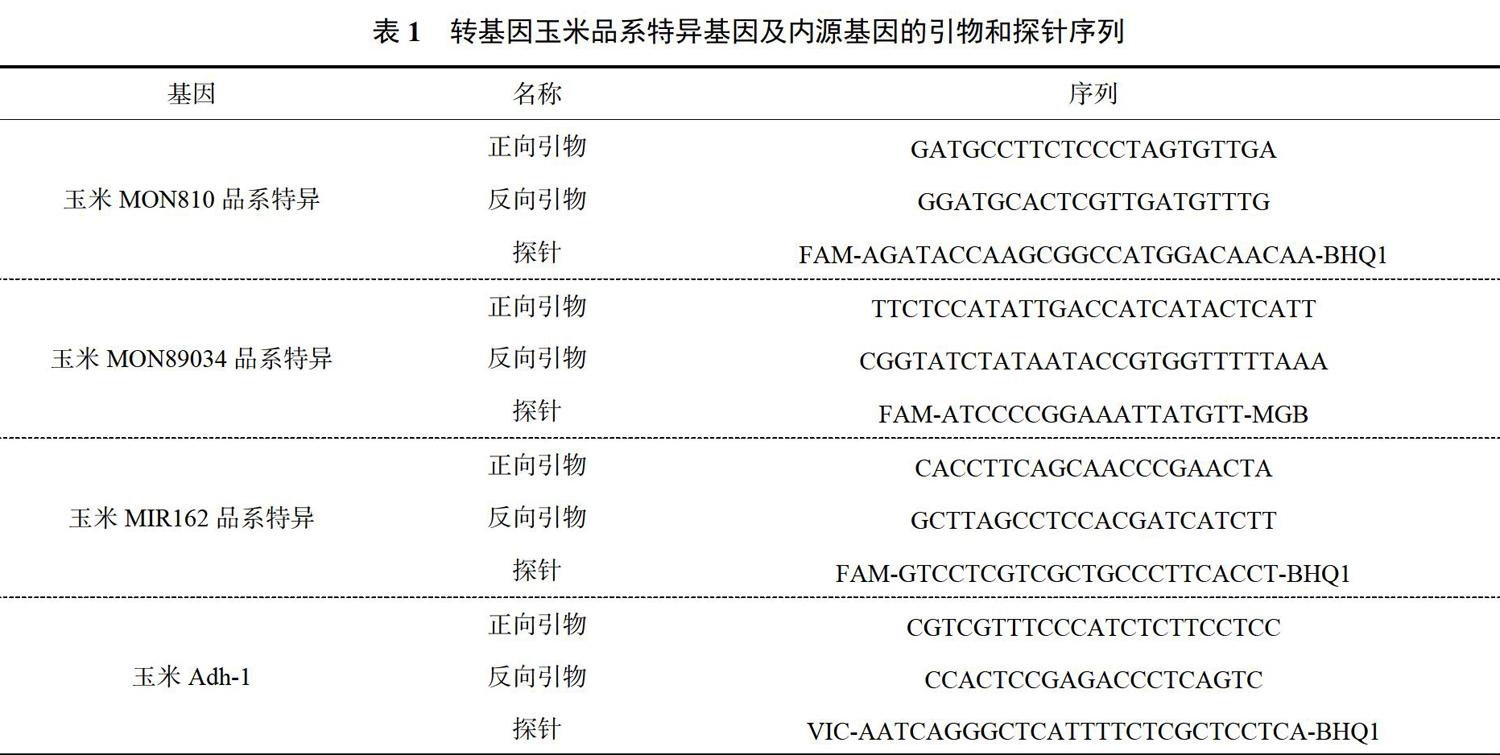
3种玉米品系的内源基因均采用Adh-1基因为检测靶基因,外源基因选择每个玉米品系的特异性序列[10-11],引物探针的序列见表1。以转基因样品MON89034为例,用实时荧光PCR反应检验设计引物探针的特异性。在含转基因MON89034外源基因的引物探针或内源基因Adh-1引物探针的PCR反应体系中,分别加入转基因玉米MIR162品系、MON810品系、玉米GA21品系、玉米TC1507品系、玉米T25品系、玉米NK603品系、转基因玉米MON89034品系和非转基因玉米的基因组DNA,验证该引物探针体系是否只针对MON89034基因组有品系特异性外源基因扩增曲线,其他样品仅有内源基因扩增。每个转基因玉米品系的芯片dPCR反应体系(总体积为20μL)中,内外源基因的正反向引物终浓度均为500nmol/L,探针终浓度为100nmol/L。Master mix体积为10μL,H2O体积为3.2μL。3D芯片dPCR的反应条件为酶激活和预变性阶段:96℃,10min;循环扩增阶段:60℃/2min,98℃/30s,共40个循环。升温速率为0.8℃/s,降温速率为1.2℃/s。
1.5 定量限和线性范围验证
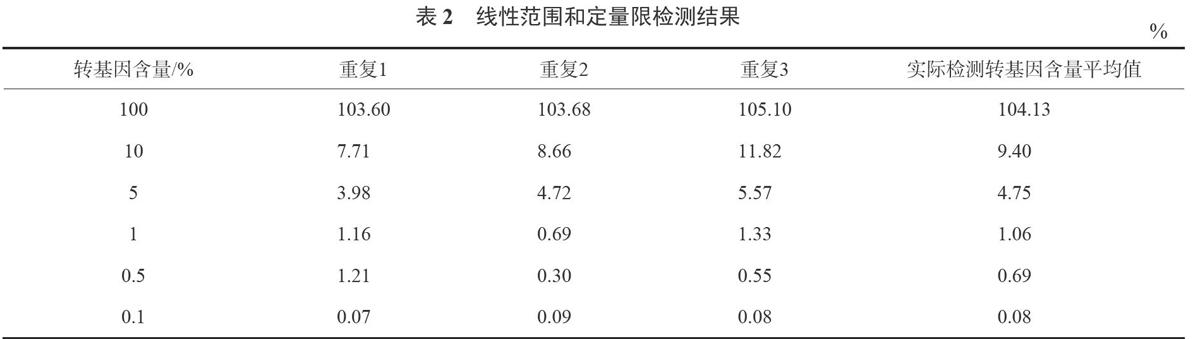
定量检测低限是指被检测的样品在合适的精度水平下(一般转基因定量检测要求RSD小于25%),能被检测出的最低含量或浓度[12]。本文将转基因含量为100%的转基因玉米品系MON89034与阴性的玉米种子粉末进行混合,制成10%、5%、1%、0.5%、0.1%的样品,验证其转基因含量检测的定量限和线性范围。
1.6 精密度实验
dPCR的实验结果是通过统计上万个微小反应单元的结果得出,重复性好。精密度实验方案为在相同的PCR反应体系中分别加入相同的基因组DNA样品(选择了1%、10%、100%转基因含量的3种样品),实验重复8次,统计8次检测结果的RSD。
1.7 玉米品系混合样品检测
采用双盲样验证法,由实验室不同人员利用1.1中转基因玉米种子粉标准物质按照质量配比配制成玉米品系混合样品,然后按照1.2中的方法提取斟建且DNA。盲样1为含25%玉米MIR162和50%玉米MON89034混合样品,盲样2为含50%玉米MIR162和4%玉米MON810混合样品,分别由转基因玉米MIR162,MON89034和MON810的扩增体系对混合DNA样品进行扩增,验证3个不同玉米品系数字PCR方法定量检测混合样本的准确性。
1.8 微滴dPCR和芯片dPCR结果的一致性验证
将1.1中转基因玉米种子粉标准物质寄送给两家不同公司,进行DNA提取实验和dPCR实验。在微滴dPCR与芯片dPCR实验中采用相同的PCR反应体系(相同的引物探针浓度,但PCR预混液Master mix不同,与仪器配套使用),选择与平台相适应的PCR反应程序进行实验,每种平台实验重复8次,用T-test比较两种方法的实验结果,判断是否具有显著性差异。
2 实验结果
2.1 反应条件的建立
本文中所设计的引物探针仅对特定玉米品系有明显的扩增曲线,对非目标玉米品系无扩增。在芯片dPCR中,所有玉米品系都同时进行外源基因和玉米内源基因的扩增。为了避免可能出现的两个靶基因的扩增体系相互干扰,本文设计了多个内源基因和外源基因的引物探针进行配对,选出内外源基因都能正常扩增且两种荧光信号彼此不干扰的扩增体系。每种转基因玉米的扩增图见图1。研究结果表明,3个转基因玉米品系的内外源基因可以在同一反应体系中进行,且阴性反应孔和阳性反应孔的荧光信号能明显区分。
2.2 线性范围和定量限检测结果
将转基因含量为100%的转基因玉米品系MON89034与阴性的玉米进行混合,制成10%、5%,1%、0.5%、0.1%的种子粉末样品。提取样品的基因组DNA并进行dPCR的扩增。每个样品进行3次重复检测,取3次检测的平均值作为定值结果。扩增结果显示,本方法检测0.1%转基因含量的种子样品时RSD为12.5%。本方法检测转基因含量0.1%及以上的样品RSD小于25%,即本方法定量限可达到0.1%。检测转基因含量标准值为0.1%~100%的种子样品,检测结果方程为Y=1.042X-0-002,其中X为转基因含量标准值,Y为转基因含量实际检测值,线性r2为0.999。具体检测结果见表2。
2.3 方法精密度检测
用转基因含量为100%、10%、1%的转基因玉米MON89034基因组DNA测试方法的精密度,重复测量8次,检测结果精密度见表3。不同转基因比例的精密度均优于10%,符合国际上转基因定量结果RSD优于25%的要求。
2.4 混合种子粉末样品检测结果
在盲样检测中,25%玉米MIR162混合样品的定量结果为19.6%,50%玉米MON89034混合样品的定量结果为66.7%,50%玉米MIR162混合样品的定量结果为64.7%,4%玉米MON810混合样品的定量结果为3_3%,定量结果的误差均小于25%,符合国家转基因定量检测的需要。
2.5 两种平台实验结果的一致性验证
dPCR主要有微滴式和芯片式两种平台,微小反应单元的形成方式有差异。微滴dPCR中微滴在PCR反应前由微滴振荡器现场生成,芯片dPCR的微孔大小在制造芯片时已经确定。微滴dPCR是在油包水的液体中进行反应,加热速度快,PCR反应中的退火温度、时间和芯片式dPCR有所区别。芯片dPCR由于芯片的影响,热传导时间较慢,因此芯片dPCR中60℃退火延伸的时间较长,需要2min。而微滴PCR在油包水的液體条件下扩增,退火温度在56℃反而能到达更好的扩增效果,扩增时间为1min,图2为微滴PCR退火温度的优化情况,MON89034品系特异基因扩增优化情况见图A,图B。图A阴性(黑色)信号值在5000~6500之间,阳性(蓝色)信号在6000~10000之间,阴性和阳性信号不能明显区分。图B优化退火温度后,阴性信号在1000~2000之间,而阳性信号在6000~8000之间,阻性和阳性信号分离情况好。内源基因Adh-1的扩增情况见图C、D。图C和图D中阴性孔的荧光值(黑色)没有变化,阳性孔的荧光信号值(蓝色)在优化后由3000提高到3500~4000,最终阴性和阳性反应孔的荧光值分离情况更好。微滴dPCR的反应条件为:酶激活和预变性阶段:95℃,10min;循环扩增阶段:56℃/1min,94℃/30s,共40个循环。
将3种转基因玉米粉末交给两家不同公司进行玉米基因组DNA的提取和dPCR检测实验。3种转基因玉米品系在两种平台的检测结果见表4。尽管由于提取DNA浓度不同,两种平台的拷贝数浓度有差异,但其转基因含量定量结果的一致性很好。转基因含量100%的转基因玉米MON89034,芯片dPCR和微滴dPCR的定量结果分别为105.6%和106.5%,将两种检测方法的结果进行T一检验,其显著性差异P=0.58>0.05,证明两种方法的检测结果没有显著性差异。转基因含量为10%的转基因玉米MON810,芯片dPCR和微滴dPCR的定量结果分别为9.7%和9.5%,显著性差异P=0.38>0.05。转基因含量100%的转基因玉米MIR162,芯片dPCR和微滴dPCR的定量结果分别为114.2%和115.8%,显著性差异P=0.39>0.05。3种转基因玉米用两种不同平台检测结果的T-检验表明,两种平台的检测结果没有显著性差异。
3 结束语
dPCR定量检测转基因产品的灵敏度高,特异性好,可以应用于更多不同转基因植物品系的定量检测。本文针对进出口贸易中涉及最多的3个转基因玉米品系MON89034,MON810,MIR162设计引物探针进行dPCR定量检测。通过对不同玉米品系的qPCR实验,验证了用于3个玉米品系检测的引物与探针的特异性。在qPCR实验中具有特异性的内外源基因的引物探针有多种,但能应用在dPCR同一个反应微小单元中,并且内外源基因的扩增体系不相互干扰却并不容易,很常见的现象是内源或外源基因的扩增受到抑制,从而阴性反应孔和阳性反应孔的荧光信号无法区分,因此不能将qPCR中的反应条件直接应用到dPCR中。本文通过多对引物探针组合,并筛选最佳引物探针浓度、退火温度、PCR反应过程的时间、温度等,从而建立一套完整的转基因玉米dPCR定量检测方法。该检测方法中,每个微反应体系都含有两套引物探针,分别用FAM和VIC荧光通道进行检测,能实现内外源基因的同时检测,避免了同一样品因取样不一致造成的定量差异,从而节约了检测成本。该方法可以同时应用到市面上最常用微滴dPCR平台和3D芯片dPCR平台。两种平台的加热介质有很大的差异,芯片dPCR是金属腔室,其微小反应单位的体积变动较小,但热传导性略慢,因此退火延伸需要的时间较长,和 qPCR实验有很大的差异。而微滴dPCR是油包水的液滴,其反应的程序和qPCR实验基本一致,这样也利于用qPCR实验进行PCR反应程序优化。但优化实验条件后两种平台的结果一致性很好,T-test中3种转基因玉米品系的转基因含量平均值和重复性RSD没有显著性差异。该方法的定量限为0.1%,转基因定量检测范围覆盖0.1%~100%,线性系数为0.999,精密度优于10%。该方法快速高效,在检测结果的准确度、稳定性优于传统的qPCRa在实验操作上,和qPCR相比不需要制作标准曲线,避免购买昂贵的标准物质,也避免了标准物质和检测样品之间的PCR扩增效率差异对实验结果的影响。将该方法用于转基因玉米的定量检测,能为规范我國转基因监管工作提供技术支撑。
参考文献
[1]TYCZEWSKA A,WOZNIAK E,GRACZJ,et al.Towardsfood security:current state and future prospects ofagrobiotechnology[J].Trends Biotechnol,2018,36(12):1219-1229.
[2]刘信.适于转基因产品检测的核酸扩增技术[J].中国农业科技导报,2011,13(6):78-81.
[3]JACCHIA S,KAGKLI DM,1」EVENS A,et al.Identificationof single target taxon-specific reference assays for the mostcommonly genetically transformed crops using digital dropletPCR[J].Food Control,2018,93:191-200.
[4]贺鹏,马静.应用于转基因食品检测的PCR技术及其进展研究[J].食品安全导则,2018,32:74-75.
[5]DANY M,DEJAN S,MOJCA M.Quantitative analysis offood and feed samples with droplet digital PCR[J].PLOSONE,2013,8(5):e62583.
[6]DEMEKE T,DOBNIK D.Critical assessment of digital PCRfor the detection and quantification of genetically modifiedorganisms[J].Anal Bioanal Chem,2018,410(17):4039-4050.
[7]KOPPEL R,BUCHER T.Rapid establishment of dropletdigital PCR for quantitative GMO analysis[J].European FoodResearch&Technology,2015,241(3):1-13.
[8]胡佳莹,姜羽,杨立桃.利用QuantStudio(TM)3D数字PCR分析转基因玉米 MON863含量[J].农业生物技术学报,2016,24(8):1216-1224.
[9]FAZEKASDE S,GROTH S T.The evaluation of limitingdilution assays[J].Journal of Immunological Methods,1982,49(2):RI 1-R23.
[10]DOBNIK D,SPILSBERG B,BOGOZALEC KOSIR A,et al.Multiplex droplet digital per protocols for quantification of gmmaize events[J].Methods Mol Biol,2018,1768:69-98.
[11]转基因成分检测玉米检测方法:SN/T 1196-2012[S].北京:中国质检出版社,2013.
[12]GRYSON N.Effect of food processing on plant DNAdegradation and PCR-based GMO analysis:a review[J].AnalBioanal Chem,2010,396(6):2003-2022.
(编辑:莫婕)
猜你喜欢
求知导刊(2016年33期)2017-01-20
东方教育(2016年21期)2017-01-17
未来英才(2016年15期)2017-01-12
黑龙江教育·高校研究与评估(2016年12期)2017-01-11
中国绿色画报(2016年7期)2016-12-26
吉林农业·下半月(2016年12期)2016-12-26
中国新技术新产品(2016年23期)2016-12-26
科技视界(2016年26期)2016-12-17
考试周刊(2016年23期)2016-05-13
