
双层石墨烯层间限域CO氧化反应的密度泛函研究*
2019-11-08 08:45崔树稳李璐魏连甲钱萍
物理学报 2019年21期
崔树稳 李璐 魏连甲 钱萍
1) (沧州师范学院物理与信息工程学院,沧州 061001)
2) (中国科学院力学研究所,非线性力学国家重点实验室,北京 100190)
3) (北京科技大学数理学院,北京 100083)
利用密度泛函理论,研究了双层石墨烯层间一氧化碳(CO)与氧(O)的氧化反应,获得了双层石墨烯层间距与反应能垒的定量关系.计算结果表明反应初态、过渡态、末态体系总能以及反应能垒对层间距离变化敏感: 随着层间距的逐渐缩小,反应能垒逐渐增加.因此,改变双层石墨烯层间间距可以实现反应能垒的原子级调控.通过差分电荷密度分析体系的电子结构,发现当双层石墨烯层间距较小时,过渡态O-C=O中碳原子与石墨烯上下层中的碳原子之间有明显的电荷堆积,出现sp轨道杂化,导致二者相互作用增强,在z轴方向受到束缚力,难以与吸附在石墨烯表面的氧原子形成较弱的O-C键,阻碍了过渡态O-C=O的形成.通过调控双层石墨烯间距,可以降低一氧化碳氧化反应能垒.该研究可为石墨烯的应用以及新型碳基插层复合材料的制备提供一定的理论支撑.
1 引 言
自从2004年英国科学家Novoselov和Geim首次从石墨中分离出石墨烯以来,它就成为备受瞩目的研究热点[1].石墨烯是一种新型碳纳米材料,以六元环为基本单位形成二维平面结构,由于其结构独特而具有优良的电学、热学等性质,在新兴材料领域具有广泛的应用,例如石墨烯的吸附性能的应用[2,3]、石墨烯用于超电容[4]和锂离子电池的电极[5].二维材料的特点是其平面内成键能力强,面间相互作用弱,相邻二维层之间或二维覆层与基体表面之间的空间受限,引起人们广泛的研究.双层石墨烯(bilayer graphene,BLG)层间以范德瓦耳斯力结合,层间区域二维空间构成微环境,分子在其内的结构和化学反应都出现了新的现象,可以看作是一个纳米反应器,具有限域作用,这为调控化学反应提供了新的途径.
1981年Verhoff等[6]首次提出限域反应的概念,他们认为当限域反应体系空间内,有限的原材料被消耗掉或可利用的空间被完全填满时,材料生长过程就会停止.基于空间限域生长的原理便可构筑纳米尺度的限域反应体系以实现限域合成,达到对材料微观结构和形貌等的控制.在研究限域反应时,由于零维结构的分子筛和一维结构的碳纳米管构型都比较复杂,利用他们的限域体系作为模型结构,研究微观尺度上的限域反应具有较大的挑战.Yao等[7]对石墨烯与金属之间的弱相互作用充分认识的基础上,创新性地提出利用石墨烯与金属表面之间形成的两维空间可以作为纳米反应器,并进行了石墨烯限域下的表面催化反应研究,研究了CO在石墨烯/铂(Pt(111))体系中的吸附、脱附过程以及石墨烯与Pt界面间的CO完全氧化反应,理论计算认为Pt(111)表面的石墨烯不会阻碍CO分子在Pt表面的吸附,CO,O2等分子在近常压条件下能够迅速插层到石墨烯与金属界面,这种由石墨烯层和金属表面形成的限域空间中独特的电子环境,降低了CO氧化反应的活化能,使催化反应速率明显加快.实验上也研究两维材料与固体表面形成的微结构,能够看作两维的纳米反应器,以及两维材料限域下的分子插层和催化反应增强等现象,说明存在两维限域催化效应[8,9].同时,石墨烯具有的层状结构与金属表面构成的两维限域空间,可以通过构建两维平板模型进行理论研究.因此两维材料限域微环境可以提供一种理想的、实用的模型系统,来探索受限反应的基本原理[10−12].Lei等[13]通过高温快速焙烧碳包覆的超薄SnO2纳米层,在石墨烯层间限域生长了Sn量子点,有效地控制了Sn量子点的颗粒尺寸,提高了Sn量子点的配位数,作为二氧化碳还原电催化剂,表现出优异的催化活性.Zhang等[14]发现当温度和压强适合时,氧气可以在石墨烯/钌(Ru(0001))界面之间发生插层,一氧化碳可以在石墨烯/镍(Ni(111))体系发生插层.已有的研究发现,在石墨烯/铂(Pt(111))模型体系中,CO,O2等小分子能够有效地进入层间并发生反应.他们从实验和理论上探索通过体系尺寸变化调控催化特性的方法和概念,提出基于限域效应的催化“尺度调控法”的原理.通过改变石墨烯表面结构改变表面电子性能,最终实现化学反应的调控.
最近,Li等[15]利用密度泛函方法研究了限域催化作用,得出两维材料与金属表面之间的范德瓦耳斯作用和两维材料覆盖层,可以改变金属表面上的势能分布,金属表面上原子在两维限域下吸附弱化,致使吸附分子的失稳.他们把这种现象类似于施加了一个外电场于固体表面上,为了说明限域微环境通过与活性结构和反应物的弱相互作用而实现对表面化学的调变作用,Li等[15]提出“限域场”的概念,这种限域效应为未来设计高效催化剂提供了重要的参考依据.
虽然石墨烯与金属表面之间形成的两维空间,作为纳米反应器方面研究很多,对于双层石墨烯层间性质的研究相关报道很少.Wang等[16]通过实验及第一原理计算得出,插入双层石墨烯层间的氢原子的一些基本迁移路径的势垒会被极大地降低(氢原子在石墨烯表面的吸附和解吸附过程,可能是石墨烯层间最简单的化学反应).周晓峰等[17]通过密度泛函理论,研究了三明治结构graphene−2Li−graphene的储氢性能.受到上面提到的表面模型体系研究结果的启发,我们希望研究双层石墨烯层间纳米通道内的限域反应.
本文利用密度泛函理论,研究了双层石墨烯层间一氧化碳(CO)与氧(O)的氧化反应,获得了双层石墨烯层间距与反应能垒的定量关系,并分析双层石墨烯的上层和下层电子结构对化学反应的影响.第二部分给出计算模型和方法,第三部分给出结果并进行讨论,最后结论在第四部分给出.
2 计算模型和方法
2.1 计算模型
双层石墨烯模型采用4 × 4 × 1超胞(共64个原子).超胞优化后的晶格常数为a=b=2.4607 Å (a和b在xy平面上),c=6.6927 Å,α=β=90°,γ=120°.c轴真空层的厚度设置为20 Å,以避免c轴方向超胞间的相互作用.
双层石墨烯结构优化后的层间距为3.4 Å.C-O键长为1.14 Å,当CO和O插入双层石墨烯层间时,全局优化得到双层石墨烯间距膨胀至5.9 Å.下表层吸附O原子后,为保证O原子距上层石墨烯间距垂直距离大于3.4 Å,双层石墨烯层间距d选取在 4.7-5.9 Å范围,选取间隔为0.3 Å.优化后整个不同层间距体系的几何结构侧面图和俯视图如图1所示(其中灰色原子标定为双层石墨烯中的C原子,金色标定CO (CO2)中的C原子,红色标定为O原子),所有的单位都是Å.
2.2 计算方法和参数设定
本文所有计算过程均基于密度泛函理论并采用VASP (Viennaab-initiosimulation package)软件包完成计算[18,19].离子实与价电子间的相互作用采用投影缀加平面波赝势[20,21],并设定相应的截断能为400 eV,交换关联项采用广义梯度近似的Perdew−Burke−Ernzerhof泛函进行描述[22];自洽场收敛标准为 1×10-5eV,原子受力收敛标准为1×10-2eV/Å,K点网格取 3×3×1 ,使K点间距为0.3 Å-1.考虑范德瓦耳斯修正(PBE + D3)[23],结构优化采用CG (conjugated gradient)算法.CO氧化反应过渡态搜寻以及能垒计算采用CINEB方法(climbing−image nudged elastic band method),用反应坐标标记反应路径,该方法已经用于研究石墨烯的限域行为[24,25].

图1 初始结构优化模型 (a)侧面图;(b)俯视图Fig.1.Side view (a) and top view (b) of the initial optimized structures about different models.
3 结果与讨论
通过密度泛函理论计算研究CO与O在双层石墨烯层间氧化反应机制.首先讨论不同层间距对CO氧化反应反应能垒的影响,然后计算电子结构进一步讨论分析石墨烯对CO氧化反应的限域作用.
3.1 双层石墨烯层间反应能垒
反应能垒的大小可以反映化学反应发生的难易程度,指分子从常态转变为容易发生化学反应的活跃状态所需要的能量,或者是反应物从初始的状态到能够参加反应所需要的能量(反应能垒又称活化能),即:
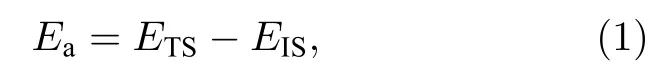
其中Ea是双层石墨烯层间一氧化碳与氧反应时的反应能垒(activation energy),EIS是反应系统初态(initial state)总能,ETS是反应系统过渡态(transition state)总能.
反应热可以写成以下形式:
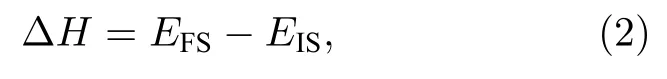
其中 ΔH是反应放出的热量,EFS是反应系统末态(final state)总能.
通过密度泛函理论计算得到双层石墨烯五个不同层间距时,氧(O)在石墨烯的顶位(top site)、桥位(bridge site)的吸附能一氧化碳与氧反应体系的反应能垒Ea和反应热ΔH,结果列于表1中,长度单位是Å,能量单位是eV.
表1 五个不同间距情况下的 (IS) ,(IS) ,Ea 和∆HTable 1.Adsorption energy at top site and bridge site,the reaction energy barrier and reaction heat at five differ−ent interlayer distances.
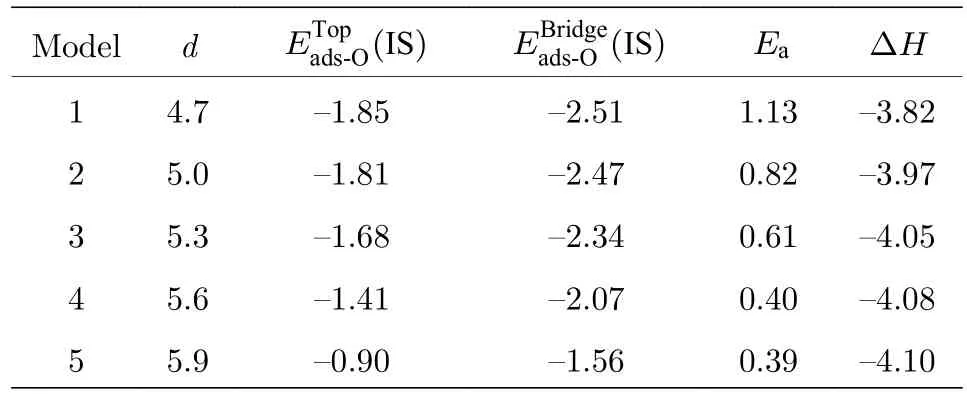
表1 五个不同间距情况下的 (IS) ,(IS) ,Ea 和∆HTable 1.Adsorption energy at top site and bridge site,the reaction energy barrier and reaction heat at five differ−ent interlayer distances.
注: d 是双层石墨烯层间距.
Modeldimages/BZ_288_1567_1866_1708_1916.pngimages/BZ_288_1795_1866_1949_1916.png images/BZ_288_2035_1878_2073_1911.png images/BZ_288_2152_1879_2214_1904.png14.7-1.85-2.511.13-3.82 25.0-1.81-2.470.82-3.97 35.3-1.68-2.340.61-4.05 45.6-1.41-2.070.40-4.08 55.9-0.90-1.560.39-4.10
由表1可得CO氧化反应为放热反应,层间距增大,释放热量增多,降低生成物所处外部环境温度,将有利于CO氧化反应动态平衡正向进行.
图2给出了反应能垒Ea与双层石墨烯层间距d之间的定量关系.从图2可以明显地看出,Ea与d呈指数关系变化,d越小,Ea越大.当d=4.7 Å时,反应能垒最大为1.13 eV,是d=5.6 Å时能垒的3倍.随着双层石墨烯层间间距的增大,反应能垒逐渐减小,关系曲线趋于平缓.其他文献也报道了相关结果,Yao等[7]计算了一氧化碳在石墨烯/Pt(111)体系中的能垒,石墨烯与Pt(111)表面间的间距d=3.3 Å,反应能垒为0.51 eV,Liu等[26]计算的结果为石墨烯与Pt(111)表面间的间距d=1.76 Å,反应能垒为0.33 eV.通过比较可以看出石墨烯与贵金属表面形成的限域环境,间距越小,能垒越小,加快反应的进行,而当二维限域的微环境为双层石墨烯时,层间距的减小增加了反应能垒,反应变慢,阻碍反应的进行.进一步说明双层石墨烯层间间距可以调控反应能垒.
图3给出了不同层间距时,各状态对应的结构图.
五个不同间距时初态、过渡态、末态下,一氧化碳中C-O键长、O与CO分子间距,以及CO2(O=C=O)中O-C与C-O键长列于表2.
我们研究了从初态IS到末态FS不同层间距离下一氧化碳氧化反应路径,具体结果如图4所示.
从图4(a)可以看出,依据过渡态理论(CINEB)计算,对应层间距为4.7 Å,双层石墨烯层间CO氧化反应能垒为1.13 eV.所涉及的反应为CO +O*=* + CO2,反应路径需要两步: 首先O*原子吸热在石墨烯内表面脱附,与CO形成过渡态O-C=O;随后快速生成CO2脱离双层石墨烯.结合表1中的反应热 ΔH,正向反应所释放的热量一部分可以满足O*脱附与CO反应所需越过势垒的能量,导致后续反应加速正向进行.然而双层石墨烯层间距缩小,则会使得上下层石墨烯对反应分子相互作用增强,令O*脱附变得困难,阻碍反应正向进行.

图2 反应能垒 Ea 与双层石墨烯层间距d的关系Fig.2.Relation between Ea and d.
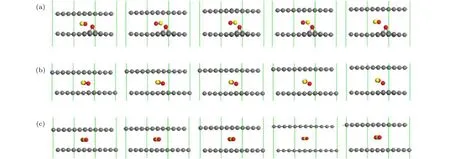
图3 各状态对应结构的侧视图 (a)初态;(b)过渡态;(c)末态Fig.3.Side view of local configurations at various states along the reaction pathway: (a) Initial state;(b) transition state;(c) final state.

表2 五个不同间距时初态(IS)、过渡态(TS)、末态(FS)的CO中C-O键长,O与CO分子间距以及CO2 (O=C=O)中O-C与C-O键长(分别对应dC-O(CO),dCO-O,dO-C(CO2),dC-O(CO2),单位为Å)Table 2.The C-O bond lengths of the initial,transition and final states of CO at five different distances,the molecular distances between O and CO,and the O-C and C-O bond lengths in CO2 (O=C=O).They correspond to dC-O(CO),dCO-O,dO-C(CO2),dC-O(CO2) with the units of Å.
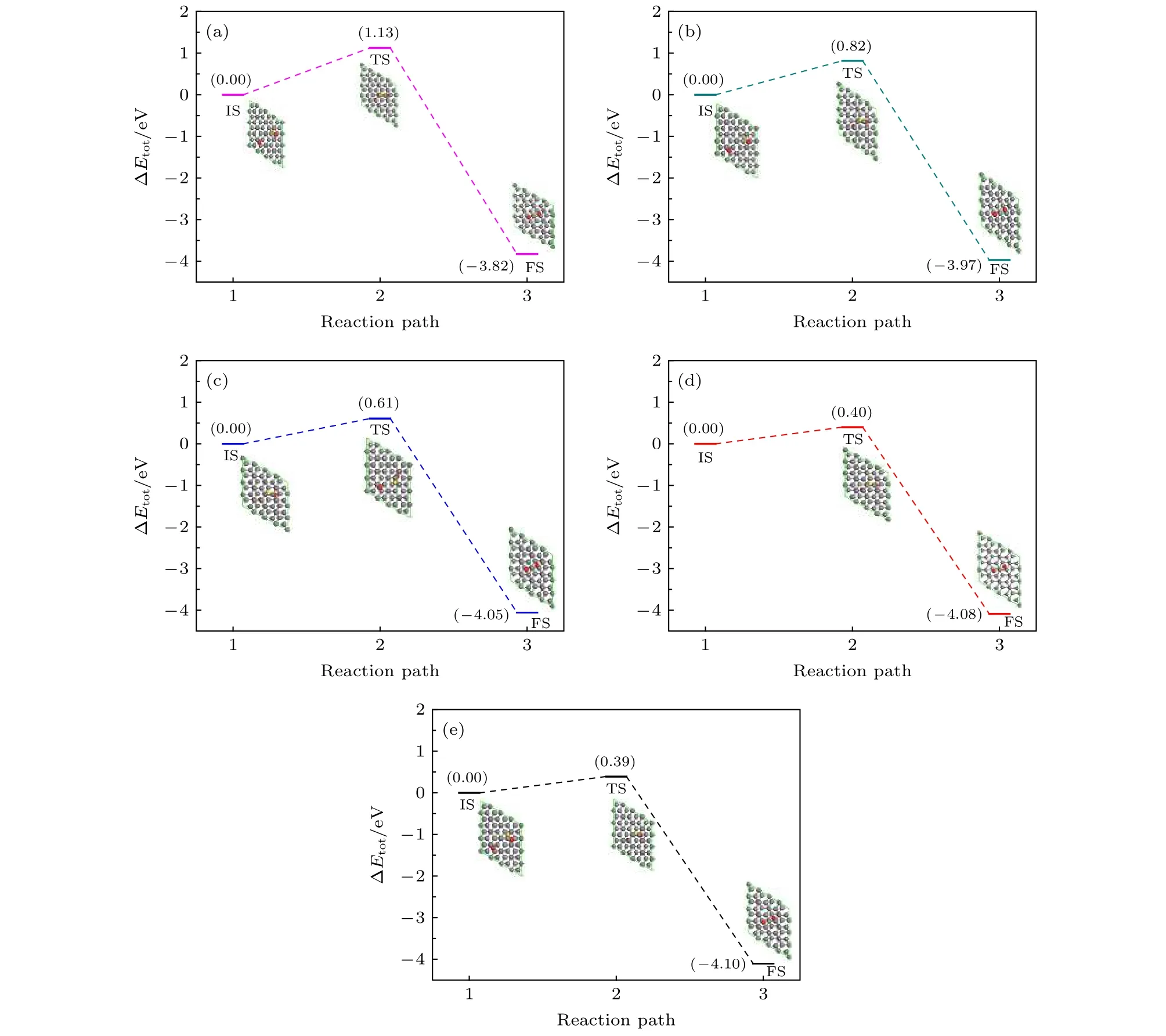
图4 不同层间距下,从初态到末态一氧化碳氧化反应路径 (a) d=4.7 Å;(b) d=5.0 Å;(c) d=5.3 Å;(d) d=5.6 Å;(e) d=5.9 ÅFig.4.Reaction pathway of the oxidation of CO and O from the initial state to final state under different interlayer distance: (a) d=4.7 Å,(b) d=5.0 Å,(c) d=5.3 Å,(d) d=5.6 Å,(e) d=5.9 Å.
3.2 差分电荷密度
为了进一步分析双层石墨烯不同层间距对一氧化碳的反应能垒的影响,探究双层石墨烯与一氧化碳、氧的电子转移,分别计算了不同层间距对应过渡态与双层石墨烯间的电荷差分密度,结果如图5所示.
图5中黄色为电子聚集区域,蓝色为电子减少区域.从图5可以看到过渡态O-C=O结构中的电荷转移情况.电子主要聚集在双层石墨上下层CO分子上方的碳原子以及原来的碳氧键中.电荷差分密度图分析表明,当层间距较大时,例如d=5.6 Å或5.9 Å时,过渡态O-C=O与石墨烯上下层之间的电荷转移较少,过渡态中的C原子与石墨烯中C原子相互作用不明显,CO与O的反应能垒较低;当层间距较小时,例如d=4.7 Å时,O-C=O与石墨烯上下层之间的电荷堆积增多,电荷转移增加,出现sp轨道杂化,导致二者相互作用增强,在z轴方向受到束缚力,难以与吸附在石墨烯表面的O原子形成较弱的O-C键,阻碍了过渡态O-C=O的形成,CO与O的反应能垒升高.较小的双层石墨烯层间距诱导s,p轨道电子局域化.当双层石墨烯靠近时过渡态(O-C=O)中C原子与上下两层石墨烯中的C原子相互作用增强,过渡态(O-C=O)在z轴方向受到束缚力,反应势垒相应增大,与计算结果一致.双层石墨烯缝隙阻碍一氧化碳与氧的氧化反应,不仅与受限空间的几何效应有关,还可能与碳材料特殊的电子结构有关.

图5 CO与O反应,在不同间距的双层石墨烯层间过渡态的电荷差分密度图,其中等值面数值分别为 (a) 0.00014 e (a.u.)3;(b) 0.00020 e (a.u.)3;(c) 0.00025 e (a.u.)3;(d) 0.00035 e (a.u.)3;(e) 0.00048 e (a.u.)3Fig.5.Electron density different of the transition states between the bilayer grapheme and the isosurface is (a) 0.00014 e (a.u.)3,(b) 0.00020 e (a.u.)3,(c) 0.00025 e (a.u.)3,(d) 0.00035 e (a.u.)3,(e) 0.00048 e (a.u.)3,respectively.
4 结 论
通过密度泛函理论研究了双层石墨烯层间一氧化碳(CO)与氧(O)的氧化反应,获得了双层石墨烯层间距与反应能垒的定量关系.计算了层间距从4.7 Å到5.9 Å时初态的能量、过渡态的能量、末态的能量以及反应能垒,当d=4.7 Å,反应能垒为1.13 eV,当d=5.9 Å,反应能垒为0.39 eV,随着层间距的缩小,反应能垒增加.为了进一步分析双层石墨烯不同层间距对一氧化碳氧化反应能垒的影响,探究双层石墨烯与一氧化碳、氧的电子转移情况,计算了对应过渡态下体系的电荷差分密度图.计算结果表明,当层间距较大时,例如d=5.6 Å或5.9 Å时,过渡态O-C=O与石墨烯上下层之间的电荷堆积少,电荷转移较少,过渡态中的C原子与石墨烯中C原子相互作用不明显,CO与O的反应能垒较低;当层间距较小时,例如d=4.7 Å时,O-C=O与石墨烯上下层之间的电荷堆积多,过渡态中的C原子与石墨烯中C原子之间存在电荷转移,形成sp轨道杂化,二者之间相互作用增强,阻碍了O-C键的形成,CO与O的反应能垒升高.较小的双层石墨烯层间距诱导s,p轨道电子局域化.当双层石墨烯靠近时过渡态(O-C=O)中C原子与上下两层石墨烯中的C原子相互作用增强,过渡态(O-C=O)在z轴方向受到束缚力,反应势垒相应增大,与计算结果一致.合适间距的双层石墨烯缝隙能显著增加CO氧化反应的能垒,进一步说明层间间距可以调控反应能垒.该研究为石墨烯的应用及新型碳基插层复合材料的制备提供一定的理论支撑.利用层间限域反应可以有效调控碳纳米材料的微观结构,并可以制备新型碳基插层复合材料,具有良好的发展前景.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中国有色金属学报(2011年4期)2011-11-08
祝您健康(1989年1期)1989-12-30
