
FBN2基因突变与遗传性结缔组织病的发生
2019-11-07 12:07徐福如蒋文君张涛姜倩张瑞雪毕宏生
遗传 2019年10期
徐福如,蒋文君,张涛,3,姜倩,3,张瑞雪,3,毕宏生,3,4
综述
基因突变与遗传性结缔组织病的发生
徐福如1,2,蒋文君2,3,张涛1,2,3,姜倩1,2,3,张瑞雪1,2,3,毕宏生1,2,3,4
1. 山东中医药大学,济南 250014 2. 山东省中西医结合眼病防治重点实验室,济南 250002 3. 山东中医药大学眼科研究所,济南 250002 4. 山东中医药大学附属眼科医院,济南 250002
遗传性结缔组织病发生后通常会累及人全身各个系统,该类疾病的发生与构成结缔组织的纤维蛋白基因发生突变引起的蛋白异构有关。原纤维蛋白-2 (fibrillin-2, FBN2)是微纤维重要组成部分,参与全身结缔组织中弹性纤维的形成。基因突变与遗传性结缔组织病,如先天性挛缩性蜘蛛指样症(congenital contractural arachnodactyly, CCA),黄斑变性(macular degeneration, MD),肌病等密切相关。研究发现基因是目前唯一已知的CCA致病基因,且不同位点的突变与CCA患者一系列临床表症的关系密切。该基因也是MD的致病基因,是肌病的风险基因,并与其他结缔组织病的发生有关。本文对基因突变引起的CCA、黄斑变性以及肌病等其他遗传性结缔组织病的临床表症及相关研究进展进行了综述,以期为深入探究基因突变致病的具体分子机制提供基础,为研究针对基因突变造成的难治性疾病的治疗药物提供理论依据。
原纤维蛋白-2;基因突变;先天性挛缩性蜘蛛样症;黄斑变性;遗传
遗传性结缔组织病的发生常与胶原蛋白和纤维蛋白出现异常密切相关,其病变会累及全身各个系统,原纤维蛋白-2 (fibrillin-2, FBN2)是微纤维重要组成部分,参与全身结缔组织中弹性纤维的形成。研究表明,基因突变会导致蛋白组装和转运障碍,引起FBN2蛋白合成减少或导致异构,影响弹性纤维的形成[1]。先天性挛缩性蜘蛛样指症(congenital contracture arachnoid, CCA)为常见遗传性结缔组织病,据美国人类遗传学中心(河滨科技)[2]DNA测序结果显示,在27%~75%的CCA患者中发现基因突变,且基因是目前唯一已知的CCA致病基因。基因突是遗传性黄斑变性(macular degeneration, MD)的致病基因,是肌病的风险基因,并与其他结缔组织疾病的发生有关。进一步明确基因突变与CCA及其他结缔组织病之间的关联,有利于相关疾病的预防、早期和产前诊断,以及精准医疗候选药物的开发。本文就基因突变引起的CCA、MD以及肌病等遗传性结缔组织病的临床表征及相关研究进展进行综述,以期为深入探究基因突变具体的分子遗传学发病机制提供基础,为研究针对基因突变造成的难治性疾病的治疗药物提供新思路。
1 FBN2结构与功能
1991年,Lee等[3]研究在MFS (Marfan syndrome)病因中的作用时,第一次分离出部分的cDNA,并通过原位杂交技术将基因定位于人类5号染色体长臂,即5q23.3。Zhang等[4]使用该部分cDNA筛选了MG-63人骨肉瘤细胞系cDNA文库,获得了全长的基因,其基因坐标为128257909~128538041,基因长度279.57 kb,包含65个外显子,转录产物为10 166 bp,mRNA包括10724个核苷酸,其中开放阅读框为8739个核苷酸,编码含2912个氨基酸残基且大小约350 kDa糖蛋白原纤维蛋白-2[5]。与FBN1结构相似,FBN2含有47个表皮生长因子样重复序列(EGF)、7个TGF-β结合蛋白重复序列(TB)、2个杂交结构域(Hyb)。但有所不同的是,FBN1和FBN2氨基末端区域分别富含脯氨酸和甘氨酸,这决定了二者在结构和功能上的差异[6]。FBN1在无FBN2时也可形成微纤维,而FBN2只有在FBN1存在时,才能形成微纤维。47个EGF样结构域中43个含钙结合保守的一致性序列,被称为cb-EGF,由6个保守的半胱氨酸(Cys)残基形成3个二硫键维持该结构域蛋白质折叠的稳定性[7,8](图1)。钙离子结合序列可以直接耦合钙离子或稳定钙离子结合位点,可避免FBN2被水解酶水解,并在FBN2与细胞外基质其他成分的相互作用中起到重要作用。TB结构域与潜伏性生长因子同源,内含8个半胱氨酸残基并形成4个二硫键,参与调节细胞外基质中TGF-β的生物活性。杂交结构域(Hyb)呈球形,由TB结构域的N末端与cb-EGF结构域的C末端融合形成,能够产生广泛的作用[9]。
以上各个结构域的完整性确保了FBN2蛋白的稳定性,从而发挥其在形成细胞外微纤维中的关键作用,并参与调节早期弹性纤维的组装过程,使结缔组织结构保持完整,进而维持人体关节的灵活和正常器官的发育及功能。基因突变引起FBN2蛋白异常表达,破坏结缔组织正常结构,最终导致全身多个器官疾病的发生。
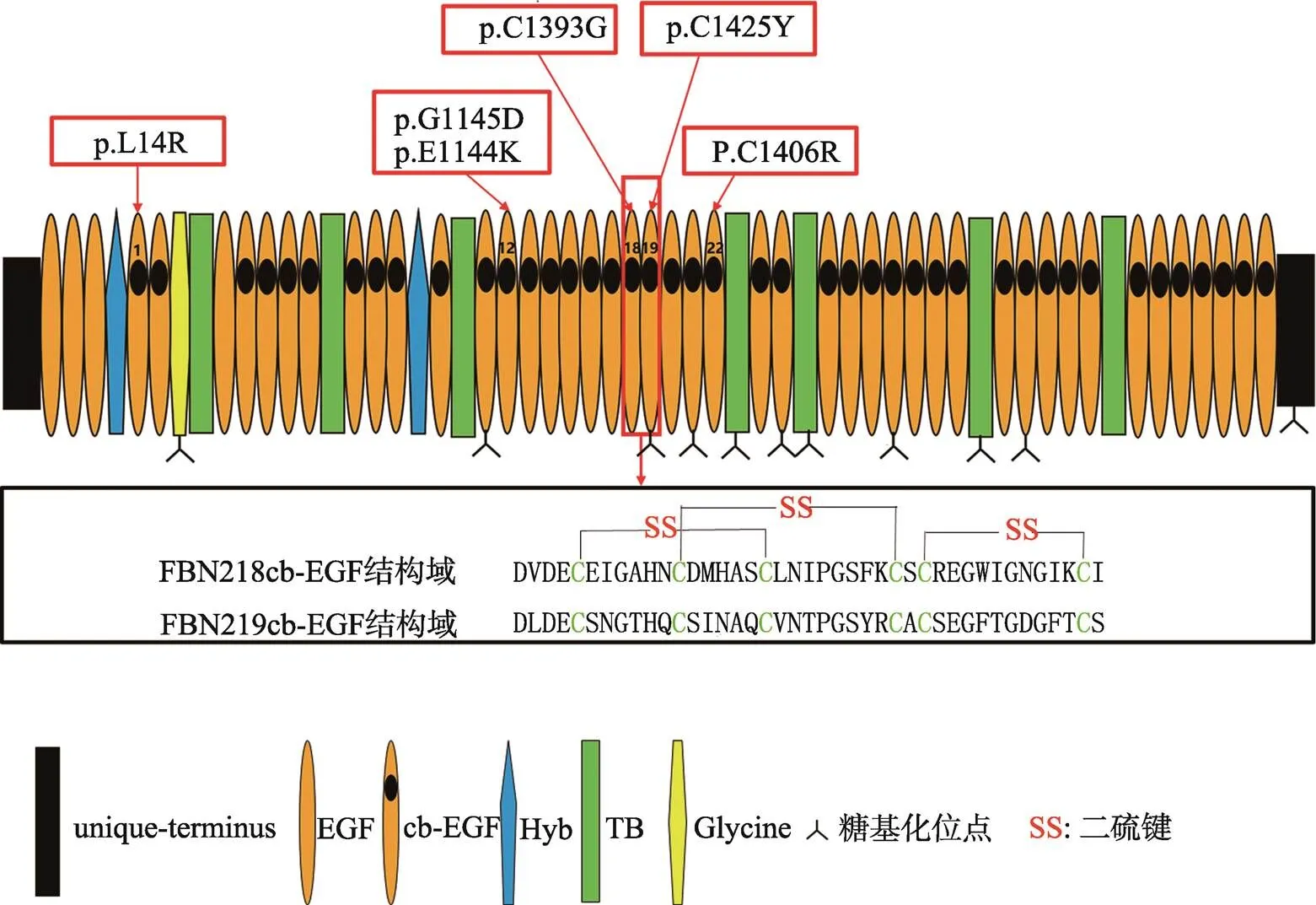
图1 FBN2蛋白结构域及突变位点
p.G1145D和p.E1144K均为第12cb-EGF结构域的突变位点;p.C1393G为第18cb-EGF结构域的突变位点;p.C1425Y为第19cb-EGF结构域的突变位点;p.C1406R为第22cb-EGF结构域的突变位点。
2 FBN2基因突变与先天性挛缩性蜘蛛样指症
CCA也称Beals综合征,是一种常染色体显性遗传的结缔组织病,临床表现多样化,其特征为蜘蛛样指、脊柱后凸,皱褶耳和肌肉发育不全等,这与MFS表征相似,但不同的是CCA患者中眼病和心脏发病率较低[10]。MFS患者常发生基因突变[11]。与基因突变相似,大多数与CCA相关的基因突变位于外显子22~36中,突变类型包括错义突变、剪接突变、片段缺失等(表1)。
2.1 错义突变与相关的临床表征
常见的基因突变类型为错义突变,会直接改变FBN2蛋白中的cb-EGF样结构域,影响细胞外基质微纤维的形成,表现为以下3种途径:(1)增加或减少cb-EGF样结构域中Cys残基,影响二硫键的形成和蛋白质的折叠;(2)改变蛋白质中的钙结合序列和结构域间序列,降低蛋白质对钙离子的结合活性,使FBN2更易被水解;(3)影响各结构域的组装,改变蛋白质的空间构象及分子间相互作用等。
2.1.1 半胱氨酸替代与相关的临床表征
基因错义突变会导致cb-EGF结构域中Cys被替代,而Cys残基在不同物种间是高度保守的。当此类突变发生后,二硫键断裂造成FBN2结构不稳定,从而引起该蛋白功能异常。先前的研究发现,此类突变主要包括外显子27中Cys1197Tyr和Gly1177Cys、外显子28中Cys1239Arg、外显子29中Cys1256Trp和Cys1267Arg等[12]。近10年间,基因新突变被不断发现和报道。Guo等[13]和Deng等[14]分别在CCA家系中发现外显子25突变c.3229T>G导致Cys1077Gly,外显子29突变c.3769T>C导致Cys1257Arg。随着基因测序技术的进步,突变所在蛋白结构域的位置被进一步确定。Liu等[15]和Zhou等[16]分别在CCA家系中发现外显子32突变c.4216T>C和c.4177T>G,分别会导致第22cb-EGF结构域中Cys1406Arg和18cb-EFG结构域中Cys1393Gly。以上基因突变的CCA家系临床症状具有多样性的特征,详见表1。而Chen等[17]在1对CCA父子中发现外显子33突变c.4274G>A导致第19cb-EGF结构域中Cys1425Tyr。其儿子在遗传了父亲的临床表征(如皱缩耳,细长指,肘膝关节挛缩症等)外,还存在其他并发症,主要表现为L5隐形脊柱裂、深髋臼窝、小腿肌肉发育不良等。
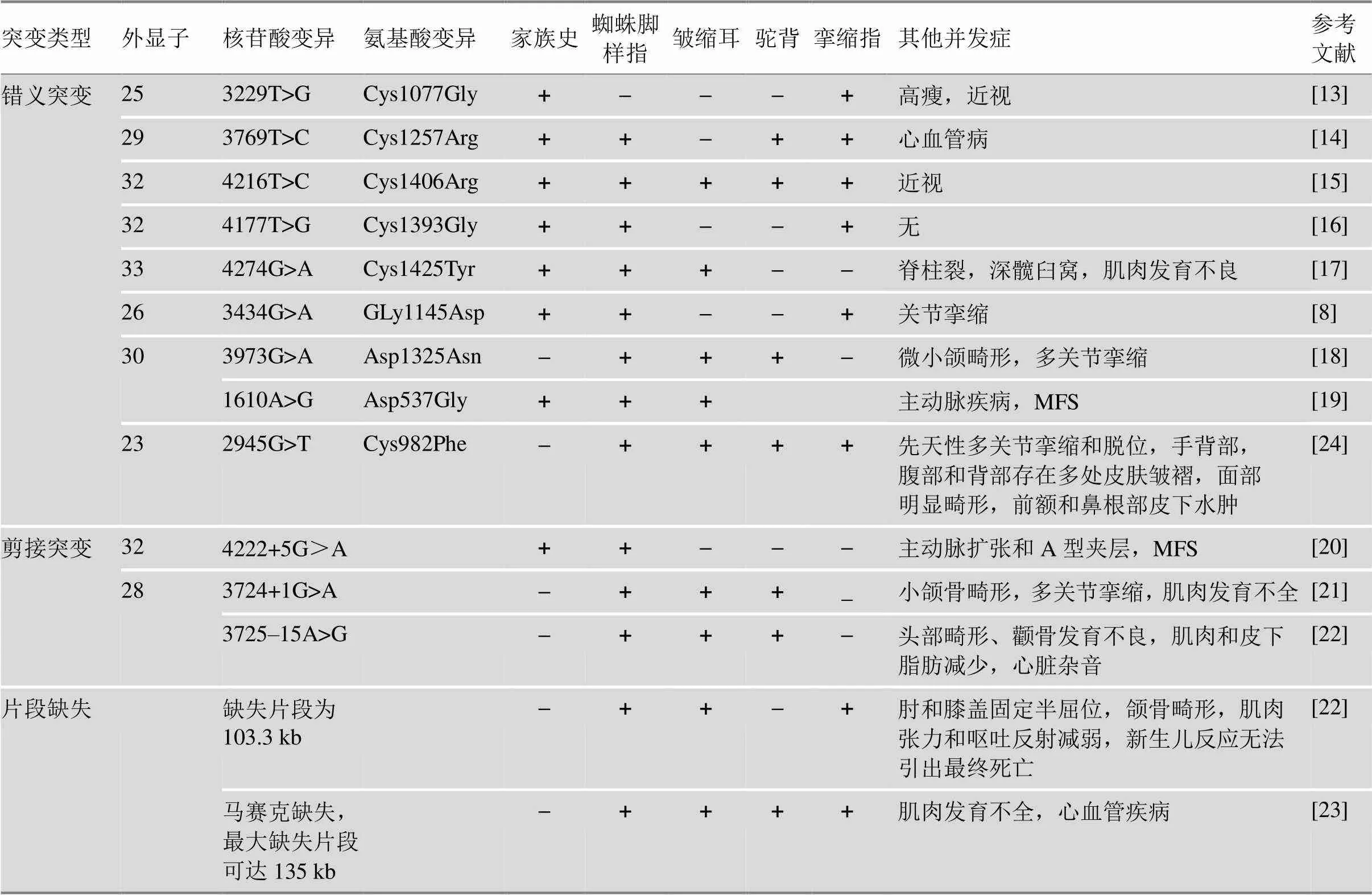
表1 不同类型的FBN2基因突变CCA患者的临床表征
“+”表示阳性;“–”表示阴性。
2.1.2 其他氨基酸替代与相关的临床表征
基因错义突变也可引起其他氨基酸替代,如天冬氨酸(Asp),天冬酰胺(Asn)以及甘氨酸(Gly)。You等[8]在中国三代CCA家系发现外显子26突变c.3434G>A导致第12cb-EGF结构域中Gly1145Asp。该突变引起亲水性氨基酸被取代,氨基酸从中性变为酸性,改变了氨基酸残基侧链构象,使相邻残基在空间发生上冲突,破坏了氢键的形成,影响蛋白质折叠结构的稳定性。此类成人患者仅出现蜘蛛脚样指、屈曲指和关节挛缩。但是,如果新生儿发生该类型突变会产生较为严重的并发症。新生儿CCA患者的外显子30发生c.3973G>A,导致p.Asp1325Asn,患儿临床表现为蜘蛛样指、多关节挛缩、耳朵皱缩、微小颌畸形和驼背等[18]。此外,Renner等[19]在CCA伴主动脉疾病的患者中发现c.1610A>G导致Asp537Gly。基因发生其他氨酸替代突变时,其病变累及全身结缔组织程度不等,会造成CCA患者不同程度的临床表征差异(表1)。
2.2 剪接突变与相关的临床表征
剪接突变会造成基因开放阅读框内发生外显子跳跃,从而影响蛋白的转录和翻译,导致FBN2蛋白结构域缩短或表达减少,引起微纤维组装障碍,进而由微纤维构成的细胞外基质结构发生紊乱以及全身结缔组织异常,最终造成全身器官疾病的发生。
Takeda等[20]在日本四代CCA家系发现c.4222+ 5G>A (IVS32+5G)A)导致外显子32跳跃,此类突变患者伴有主动脉扩张和A型夹层的严重并发症,且出现马凡式体位。Mehar等[21]在一例新生儿CCA中发现c.3724+1G>A,该突变破坏剪接位点的共有序列,计算机模拟预测显示外显子28跳跃。该患儿出生时,表现出小颌骨和外耳畸形、先天性多关节挛缩、双侧蜘蛛样指/趾、胸腰段脊柱侧凸、肌肉发育不全等症状。但患儿父母健康,基因型正常,说明该突变是新发生的,这更加确定基因突变是CCA的致病原因。另外,Inbar-Feigenberg等[22]在一例新生儿CCA患者中鉴定出基因致病突变c.3725-15A>G,该患儿除典型的症状外还存在心血管疾病和肌肉病变(表1)。
综上所述,随着对CCA遗传病因的深入研究,在临床表征各异的CCA患者中不断地鉴定出基因不同类型的新突变。此外,基因修饰、环境因素及突变都可能与CCA临床表征有关,CCA家系间和家系内相同突变个体的临床表征也存在差异,表明通过基因型无法预测到CCA患者所有临床表症,这增加了CCA的诊治难度。
2.3 片段缺失和杂合变异与相关的临床表征
基因的片段缺失可导致新生儿死亡,单个或两个基因突变引起新生儿全身器官发育异常,出现严重并发症甚至危及生命。Inbar-Feigenberg等[21]在胎儿产前羊水细胞微阵列分析中检测到基因有103.3 kb的缺失,出生后新生儿具有典型CCA临床表症外,还出现颌骨和外耳畸形,而且肌肉体积减少,肌肉张力差和呕吐反射减弱,新生儿反应无法引出,患儿于出生后第7天死亡。Laillaureix等[23]报道了第一例引起新生儿致死的基因马赛克缺失,其中缺失最小段约116 kb (chr.5:127663810~127779670),最大段135 kb (chr.5:127656238~127791139),后者包括外显子7-34 (chr.5:127656238~127791139)。该新生儿出现多发性屈曲挛缩,蜘蛛样指,耳朵皱缩,肌肉发育不全和心血管疾病等。基因内缺失发生在“新生儿(neonatal)”区域(即外显子23~34,在基因中该区域的突变可导致新生儿严重的MFS),或者阅读框架内。基因内缺失可能涉及整个基因,严重破坏微纤维组装,引发新生儿出现致死性CCA症状。Aggarwal等[24]在29周胎儿脐带血中发现致病性和双杂合子变异,突变位于基因的外显子49c.6004C>T,导致p.Pro2002Ser,基因突变位于外显子23c.2945G>T,导致p.Cys982Phe。该混合变异引起患儿临床表症与Beals和MFS的表型重合(表1),患儿在分娩时死亡。
新生儿突变往往比较严重,有致死地可能性,必要时应给予父母遗传咨询,提供产前分子诊断。CCA作为罕见的常染色体显性疾病影响后代几率为50%,应给予父母遗传咨询,疑似病例可通过羊膜穿刺术或绒毛膜绒毛取样进行产前检查并提供分子诊断,来检测家族性基因突变。
3 FBN2基因突变与其他遗传性结缔组织病
3.1 FBN2基因突变与黄斑疾病
研究表明FBN2蛋白位于Bruch膜,并在年龄相关性黄斑变性(age-related macular degeneration, AMD)眼中表达减少,在10 337例ADM患者和11 174例对照组中鉴定出一个常见基因的非同义变异体rs154001c.2893G>A,导致p.Val965Ile,该突变与AMD有关联(OR=1.10; p=3.79×10–5)[7]。Maheswar等[25]在有玻璃膜疣表型的AMD发现基因罕见错义突变c.976G>A (p.Pro326Ser)和c.4141G>T (p.His1381Asn),前者为良性,后者具有致病性,表明该基因与ADM有关。Ratnapriya1等[7]在两代常染色体显性遗传的MD家系中鉴定出基因突变c.3430G>A,导致第12cb-EGF结构域中p.Glu1144LyS。因Ca2+结合腔的负电荷有助于维持蛋白质折叠结构的稳定性并能够保证两个相邻cb-EGF结构域的相对取向,而该突变使Ca2+结合腔引入正电荷,降低了钙结合的亲和力,使相邻cb- EGF的相对取向发生顺时针改变,进而影响FBN2蛋白结构的稳定性。FBN1和FBN2聚合成10 nm微纤维,位于Bruch膜中心层,其两侧为胶原蛋白。Bruch膜能够阻止脉络膜毛细血管中的代谢物到达视网膜色素上皮层,当Bruch膜超微结构改变时,造成沉积物积累。AMD患者发生基因变异使眼组织中蛋白表达减少或丧失,影响了弹性蛋白纤维的包装,改变了柔韧性、渗透性,从而破坏了Bruch膜的屏障特性,加速脂质、免疫复合物和其他碎片的累积。另外,Shi 等[26]在Fbn2–小鼠眼睛表型中发现,与正常小鼠相比,Fbn2成年小鼠瞳孔膜微纤维排列紊乱,瞳孔不能正常回缩,悬韧小带分布不均匀,表现为在晶体赤道部分布较多,并影响附着部位晶体纤维细胞的分化。这说明基因在小鼠眼的发育过程中也发挥着关键作用,未来小鼠可用作研究基因在黄斑变性的作用的动物模型。
MD受年龄、性别、遗传等多种因素复合作用的影响,其中,基因罕见和常见变异可导致MD疾病的发生,但目前基因突变在MD患者报道不多见,随基因检测技术的提高以及患者对视力的需求的提升,基因突变在MD病因机制中的作用仍需进一步探究。
3.2 FBN2突变与肌病
遗传性肌肉变性疾病显著影响健康且伴有多种症状,如肌肉无力,心肌病和或精神发育迟滞。Dorota等[27]在一例13.5岁的严重肌无力患儿中发现基因剪切突变c.41T>G导致p.Leu14Arg。患儿父母和兄弟基因均为杂合子,而他的基因却发生纯合变异。Leu14位于第一个EGF样结构域前110残基N末端序列中,28残基-N末端是保证蛋白质正确分泌所需的信号肽,该突变破坏了该信号识别区域,显著影响蛋白质的正确分泌,导致细胞外微纤维中FBN2蛋白水平异常,进而影响弹性纤维弹性和延伸,导致肌肉发育异常。该患儿全身皮肤可见明显花纹,伸肌出现严重肌肉无力,腿部和臀部肌肉轻度无力,且脊柱前凸,马蹄足,膝关节伸直困难,频繁髌骨脱位,躯干弯曲,容易疲劳且劳累时会呼吸困难。
研究还发现小鼠基因位于18号染色体编码含2907个氨基酸的FBN2蛋白,与人FBN2蛋白具有97%的同源性[28],因此小鼠可作为研究该基因的作用的动物模型。乙酰基亚硝基脲(ENU)诱变是揭示影响肌肉发育和功能的突变有效工具。Gaynor等[29]研究发现ENU诱变引起小鼠发生纯合的Mariusz变异中携带基因的无义突变c.14T>A导致终止密码子(TAG)替换亮氨酸(TTG)的密码子,FBN2蛋白2907个氨基酸缩短到620个氨基酸,造成小鼠严重肌无力表型。此外,Gerhard 等[30]在新生Fbn2/小鼠中发现,白色脂肪异常堆积,肌肉组织出现严重缺陷,发生呼吸衰竭死亡。成年Fbn2小鼠肌酸激酶水平升高,表现为持续肌无力,与p.Leu14Arg突变导致的临床表症相似。
综上所述,FBN2蛋白能够维持肢体运动强度的肌肉结构的完整性,临床和动物模型研究结果表明基因突变是导致肌病发生的风险基因,未来仍需进一步研究基因突变在肌无力的发病的作用,为研究治疗手段提供新思路。
3.3 其他
除了上述疾病之外,基因突变还与其他结缔组织病的发生相关。2017年,袁帅等[31]在1例典型Stanford B型主动脉夹层患者中发现基因突变c.6833C>T (p.T2278),该变异为家族性胸主动脉瘤和主动脉夹层高度可疑的致病突变。2019年,Todhunter等[32]在芬兰人群中,通过队列研究发现携带FBN2rs331069 (T>C)多态变异体的受试者患动脉粥样硬化血栓性疾病比例高。提示基因多态性可能是心血管疾病的危险因素。2014年,Khoury等[33]使用TaqMan分析135例跟腱病(AT)和141例前十字韧带破裂(ACL)患者以及对照组的rs331079变异体,显示基因内含子7内的rs331079与AT和ACL破裂密切相关。Buchan等[34]在严重脊柱侧突的患者中发现基因的罕见变异,变异体位于整个基因,包括CCA患者中基因常发生变异的外显子24-34区域,如p.Arg1021Cys、p.Ile1116Ser、p.Leu1125Val以及p.Gly1271Ala。股动脉瘤患者[35]基因测序中发现基因中的杂合子变体c.8506A>G。张建华等[36]研究还发现基因甲基化与食管癌的发生发展密切相关,可以作为一种潜在的抑癌基因。
4 结语与展望
FBN2蛋白广泛存在于结缔组织的细胞外基质中,基因突变可引起结缔组织异常并累及全身各个系统。近年来CCA患者中新的基因突变位点不断被报道,该基因突变类型及位点的差异与CCA患者不同的临床表征密切相关。此外,基因突变还被发现是MD的致病基因,并且与肌病以及其他结缔组织病相关。
目前已经发现的与遗传性结缔组织病有关的突变位点覆盖人群尚少,且缺少基因突变与不同临床表征关系的深入研究。在今后的研究中,可利用二代和三代基因测序技术在更大的患者人群中鉴定基因新突变位点,并明确突变与相应疾病表型的对应关系,对实现早期产前诊断和预测个体疾病的发展规律具有重要意义。另外,基因突变在遗传性结缔组织病中致病的分子机制亟代探讨,未来可利用动物和细胞模型进一步研究FBN2蛋白与其他分子间相互的作用机制,深入理解基因的功能,并为将来实现靶向干预以及思考新的治疗方案提供思路
[1] Davis MR, Summers KM. Structure and function of the mammalian fibrillin gene family: implications for human connective tissue diseases., 2012, 107(4): 635–647.
[2] Center for Human Genetics, DNA test descriptions & CPT codes, http://chginc.org/dna-test-descriptions- cpt-codes/.
[3] Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, Tsipouras P, Ramirez F, Hollister DW. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes.,1991, 352(6333): 330–334.
[4] Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices., 1994, 124(5): 855–863.
[5] Frédéric MY, Monino C, Marschall C, Hamroun D, Faivre L, Jondeau G, Klein HG, Neumann L, Gautier E, Binquet C, Maslen C, Godfrey M, Gupta P, Milewicz D, Boileau C, Claustres M, Béroud C, Collod-Béroud G. Thegene: new mutations, locus-specific database (universal mutation database fbn2), and genotype-phenotype correlations.,2010, 30(2): 181–190.
[6] Trask TM, Ritty TM, Broekelmann T, Tisdale C, Mecham RP. N-terminal domains of fibrillin 1 and fibrillin 2 direct the formation of homodimers: a possible first step in microfibril assembly.,1999, 340(Pt 3): 693– 701.
[7] Ratnapriya R, Zhan X, Fariss RN , Branham KE , Zipprer D, Chakarova CF, Sergeev YV, Campos MM, Othman M, Friedman JS, Maminishkis A, Waseem NH, Brooks M, Rajasimha HK, Edwards AO, Lotery A, Klein BE, Truitt BJ, Li B, Schaumberg DA, Morgan DJ, Morrison MA, Souied E, Tsironi EE, Grassmann F, Fishman GA, Silvestri G, Scholl HP, Kim IK, Ramke J, Tuo J, Merriam JE, Merriam JC, Park KH, Olson LM, Farrer LA, Johnson MP, Peachey NS, Lathrop M, Baron RV, Igo RP Jr, Klein R, Hagstrom SA, Kamatani Y, Martin TM, Jiang Y, Conley Y, Sahel JA, Zack DJ, Chan CC, Pericak-Vance MA, Jacobson SG, Gorin MB, Klein ML, Allikmets R, Iyengar SK, Weber BH, Haines JL, Léveillard T, Deangelis MM, Stambolian D, Weeks DE, Bhattacharya SS, Chew EY, Heckenlively JR, Abecasis GR, Swaroop A. Rare and common variants in extracellular matrix gene fibrillin 2 (FBN2) are associated with macular degeneration.,2014, 23(21): 5827–5837.
[8] You G, Zu B, Wang B, Wang Z, Xu Y, Fu Q. Exome sequencing identified a novel FBN2 mutation in a Chinese family with congenital contractural arachnodactyly.,2017, 18(4): 626–635.
[9] Jensen SA, Iqbal S, Lowe ED, Redfield C, Handford PA. Structure and interdomain interactions of a hybrid domain: a disulphide-rich module of the fibrillin/tlbp superfamily of matrix proteins.,2009, 17(5): 759–768.
[10] Meena JP, Gupta A, Mishra D, Juneja M. Beals-hecht syndrome (congenital contractural arachnodactyly) with additional craniospinal abnormality: a case report.,2015, 24(3): 226–229.
[11] Chen QQ, Wu YA, Huang XL, Chen T, Huang Y, Chen FL, Chen FW. Analysis of two new mutations ingene in Han people with Marfan syndrome (MFS)., 2010, 32(1): 49–53.陈清泉, 伍严安, 黄肖利, 陈同, 黄毅, 陈发林, 陈发文. 汉族马凡综合征(MFS)患者基因两种新发突变分析. 遗传, 2010, 32(1): 49–53.
[12] Gupta PA, Putnam EA, Carmical SG, Kaitila I, Steinmann B, Child A, Danesino C, Metcalfe K, Berry SA, Chen E, Delorme CV, Thong MK, Adès LC, Milewicz DM. Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype., 2002, 19(1): 39–48.
[13] Guo X, Song C, Shi Y, Li H, Meng W, Yuan Q, Xue J, Xie J, Liang Y, Yuan Y, Yu B, Wang H, Chen Y, Qi L, Li X. Whole exome sequencing identifies a novel missense FBN2, mutation co-segregating in a four-generation chinese family with congenital contractural arachnodactyly,2016, 17(1): 91.
[14] Deng H, Lu Q, Xu H, Deng X, Yuan L, Yang Z, Guo Y, Lin Q, Xiao J, Guan L, Song Z. Identification of a novel missense FBN2 Mutation in a chinese family with congenital contractural arachnodactyly using exome sequencing.,2016, 11(5): e0155908.
[15] Liu W, Zhao N, Li XF, Wang H, Sui Y, Lu YP, Feng WH, Ma C, Han WT, Jiang M. A novel FBN2 mutation in a chinese family with congenital contractural arachnodactyly, 2015, 5: 163–166.
[16] Zhou S, Wang F, Dou Y, Zhou J, Hao G, Xu C, Wang QK, Wang H, Wang P. A novel mutation cosegregates with congenital contractural arachnodactyly in a five-generation chinese family.,2018, 6(8): 1612–1617.
[17] Chen Y, Lei YP, Zheng HX, Wang W, Cheng HB, Zhang J, Wang HY, Jin L, Li H. A novel mutation (C1425Y) in the FBN2 gene in a father and son with congenital contractural arachnodactyly., 2009, 13(3): 295–300.
[18] Gürler AI, Yüksel Z, Karaer K. A novel FBN2 mutation in a turkish case with congenital contractural arachnodactyly.,2018, 27(3): 109–111.
[19] Renner S, Schüler H, Alawi M, Kolbe V, Rybczynski M, Woitschach R, Sheikhzadeh S, Stark VC, Olfe J, Roser E, Seggewies FS, Mahlmann A, Hempel M, Hartmann MJ, Hillebrand M, Wieczorek D, Volk AE, Kloth K, Koch-Hogrebe M, Abou Jamra R, Mitter D, Altmüller J, Wey-Fabrizius A, Petersen C, Rau I, Borck G, Kubisch C, Mir TS, von Kodolitsch Y, Kutsche K, Rosenberger G. Next-generation sequencing of 32 genes associated with hereditary aortopathies and related disorders of connective tissue in a cohort of 199 patients,2019. doi: 10.1038/s41436-019-0435-z
[20] Takeda N, Morita H, Fujita D, Inuzuka R, Taniguchi Y, Imai Y, Hirata Y, Komuro I. Congenital contractural arachnodactyly complicated with aortic dilatation and dissection: case report and review of literature.,2015, 167(10): 2382–2387.
[21] Mehar V, Yadav D, Kumar R, Yadav S, Singh K, Callewaert B, Pathan S, De Paepe A, Coucke PJ. Congenital contractural arachnodactyly due to a novel splice site mutation in thegene.,2014, 03(03): 163–166.
[22] Inbar-Feigenberg M, Meirowitz N, Nanda D, Toi A, Okun N, Chitayat D. Beals syndrome (congenital contractural arachnodactyly): prenatal ultrasound findings and molecular analysis.,2015, 44(4): 486– 490.
[23] Lavillaureix A, Heide S, Chantot-Bastaraud S, Marey I, Keren B, Grigorescu R, Jouannic JM, Gelot A, Whalen S, Héron D, Siffroi JP. Mosaic intragenic deletion of FBN2 and severe congenital contractural arachnodactyly., 2017, 92(5): 556–558.
[24] Aggarwal S, Das Bhowmik A, Tandon A, Dalal A. Exome sequencing reveals blended phenotype of double heterozygous FBN1 and FBN2 variants in a fetus., 2018, 61(7): 399–402.
[25] Duvvari MR, van de Ven JP, Geerlings MJ, Saksens NT, Bakker B, Henkes A, Neveling K, del Rosario M, Westra D, van den Heuvel LP, Schick T, Fauser S, Boon CJ, Hoyng CB, de Jong EK, den Hollander AI. Whole exome sequencing in patients with the cuticular drusen subtype of age-related macular degeneration., 2016, 11(3): e0152047.
[26] Shi Y, Tu Y, Mecham RP, Bassnett S. Ocular phenotype of fbn2-null mice., 2013, 54(12): 7163–7173.
[27] Monies D, Maddirevula S, Kurdi W, Alanazy MH, Alkhalidi H, Al-Owain M, Sulaiman RA, Faqeih E, Goljan E, Ibrahim N, Abdulwahab F, Hashem M, Abouelhoda M, Shaheen R, Arold ST, Alkuraya FS. Autozygosity reveals recessive mutations and novel mechanisms in dominant genes: implications in variant interpretation., 2017, 19(10): 1144–1150.
[28] Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils., 1995, 129(4): 1165–1176.
[29] Miller G, Neilan M, Chia R, Gheryani N, Holt N, Charbit A, Wells S, Tucci V, Lalanne Z, Denny P, Fisher EM, Cheeseman M, Askew GN, Dear TN. ENU mutagenesis reveals a novel phenotype of reduced limb strength in mice lacking fibrillin 2.,2010, 5(2): e9137.
[30] Sengle G, Carlberg V, Tufa SF, Charbonneau NL, Smaldone S, Carlson EJ, Ramirez F, Keene DR, Sakai LY. Abnormal activation of BMP signaling causes myopathy in fbn2 null mice., 2015, 11(6): e1005340.
[31] Yuan S, Xing XB, Xu SW, Jiang YR, Guo Y, Wang F, Zhao WN, Liu FS. A typical Stanford type B aortic dissection patient carries a FBN2 gene p.T2278M mutation in 1 case.,017, 33(12): 1237– 1239.袁帅, 刑晓博, 徐晟伟, 姜玉瑞, 郭勇, 王芳, 赵雯娜, 刘福颂. 典型Stanford B型主动脉夹层患者携带FBN2基因p.T2278M突变1例. 临床心血管病杂志, 2017, 33(12): 1237–1239.
[32] Kunnas T, Solakivi T, Nikkari ST. Gene polymorphisms of fibronectin rs2289202 and fibrillin 2 rs331069 associate with vascular disease, the TAMRISK study., 2018, 8(1): 65–68.
[33] Khoury LE, Posthumus M, Collins M, Collins M, van der Merwe W, Handley C, Cook J, Raleigh SM. ELN and FBN2 gene variants as risk factors for two sports-related musculoskeletal injuries., 2015, 36(4): 333–337.
[34] Buchan JG, Alvarado DM, Haller GE, Cruchaga C, Harms MB, Zhang T, Willing MC, Grange DK, Braverman AC, Miller NH, Morcuende JA, Tang NL, Lam TP, Ng BK, Cheng JC, Dobbs MB, Gurnett CA. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis.,2014, 23(19): 5271–5282.
[35] Ratschiller T, Müller H, Schachner T, Fellner F, Sulzbacher G, Zierer A. Femoral artery aneurysm repair in a patient with a fibrillin-2 mutation.,2018, 52(7): 583–586.
[36] Zhang JH, Guo Q, Zhou JZ, Zhang Y. Study on the correlation between FBN1/FBN2 gene methylation and esophageal carcinoma.,2018, 26(4): 523– 526.张建华, 郭琪, 周章剑, 张毅. FBN1、FBN2基因甲基化与食管癌的相关性研究. 现代肿瘤医学, 2018, 26(4): 523–526
Fibrillin-2 gene mutations associated with hereditary connective tissue diseases
Furu Xu1,2, Wenjun Jiang2,3, Tao Zhang1,2,3, Qian Jiang1,2,3, Ruixue Zhang1,2,3, Hongsheng Bi1,2,3,4
Fibrillin-2 (FBN2) is an important component of microfibers which are involved in the formation of elastic fibers in connective tissue throughout the human body. Hereditary connective tissue diseases may result from genetic mutations ofcausing heterogeneity of fibrin. Genetic mutations of FBN2are associated with a variety of hereditary connective tissue diseases including Congenital Contractural Arachnodactyl (CCA), Macular Degeneration (MD), and myopathy. Studies have shown that thegene is recognized as the only pathogenic gene related to CCA and that CCA patients have different clinical presentations depending on the identified genetic mutations at differentsites. In this review, we summarize the roles of, its mutations and impact on the physiological and pathological processes of many hereditary connective tissue diseases. We include brief descriptions of clinical manifestations of these diseases providing a basis for further exploration of the specific molecular mechanism ofgene mutation pathogenesis which provides a theoretical basis for the therapy and medications for refractory diseases caused bygene mutation.
fibrillin-2; gene mutation; congenital contracture arachnoid; macular degeneration; genetic
2019-04-08;
2019-05-30
国家自然科学基金项目(编号:81603421),山东省重点研发计划项目(编号:2016GGH3119)和山东省重点研发计划项目(编号:2017CXGC1211)资助[Supported by the National Natural Science Foundation of China (No.81603421), Shandong Province Key Research and Devlepment Program (No.2016GGH3119), Shandong Province Key Research and Devlepment Program (No.2017CXGC1211) ]
徐福如,在读硕士研究生,专业方向:屈光不正及白内障。E-mail: xufuru1995@163.com
毕宏生,博士,教授,研究方向:屈光不正及白内障。E-mial: hongshengbi@126.com
10.16288/j.yczz.19-052
2019/6/4 15:32:38
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190604.1532.004.html
(责任编委: 吴志英)
猜你喜欢
中国现代医生(2022年19期)2022-11-04
电子科技大学学报(2022年5期)2022-10-29
昆明医科大学学报(2022年4期)2022-05-23
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年2期)2021-01-18
中国生殖健康(2018年4期)2018-11-06
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
现代检验医学杂志(2016年4期)2016-11-15
西南军医(2015年1期)2015-01-22
