
常染色体隐性遗传小头畸形相关蛋白研究进展
2019-11-07 12:06王玉杰周小坤徐丹
遗传 2019年10期
王玉杰,周小坤,徐丹
综述
常染色体隐性遗传小头畸形相关蛋白研究进展
王玉杰,周小坤,徐丹
福州大学生物科学与工程学院,福州 350108
脑发育相关疾病是一类影响大脑或中枢神经系统生长和发育的疾病。常染色体隐性遗传小头畸形(autosomal recessive primary microcephaly, MCPH)是一种神经系统发育障碍疾病,病人主要表现为头围减小,并伴随一定程度的智力衰退。迄今为止已发现至少有25个基因突变都会导致MCPH,根据它们发现的顺序分别命名为~。MCPH蛋白作为重要的成份参与调控大脑发育相关信号通路。本文对目前发现的25个MCPH相关蛋白的表达模式、细胞定位、分子生物学功能、表型及动物模型进行了综述,旨在提升人们对脑发育相关疾病的致病机制的认知,促进对神经元生成、脑尺寸大小及脑功能调控等分子机制的研究。
小头畸形;表达模式;细胞组分;表型概述;分子生物学功能;动物模型
大脑是人体最重要和最复杂的器官,拥有上千亿个神经元和彼此间相互作用形成的百万亿个连接点。大脑通过形成庞大而复杂的神经网络控制着其他器官或系统的正常功能。理解大脑的运转机制,是人类与科学届面临的最伟大的挑战之一。脑科学是21世纪各种前沿科学中最为引人关注的领域之一,同时也是多学科交叉的重要前沿科学领域。2013年,美国启动“脑计划”旨在探索人类大脑工作机制,绘制脑活动全图,并最终开发出针对大脑疾病的疗法。2018年以来,“北京脑科学中心”和“上海脑科学中心”相继成立,标志着“中国脑计划”也正式拉开了帷幕。
脑发育异常会导致其功能异常,并最终导致严重的神经疾病如自闭症、精神分裂症及小头症等[1~7]。小头症即小头畸形(microcephaly)是一种比较罕见的大脑疾病,是由于大脑神经系统发育障碍导致的,发病率在2~12/万[8]。胎儿出生后的头围测量是诊断小头症最常用的方式之一[8]。诊断小头症的临床标准是病人的头围相对于其同年龄与性别的平均值明显小2个标准差以上[9]。通常在孕28周左右应用超声波技术和核磁共振扫描检测即可发现患儿的头围测量值及脑容量低于正常同龄胎儿。小头症的主要表现是脑的重量明显轻于正常、脑回过小或根本无脑回、大脑的发育明显迟缓,甚至在婴儿第3~5个月时就停止发育,最后导致患儿的头顶变得小而尖、鼻梁凹陷、耳大、下额后缩、前额狭小并且头围明显小于正常婴儿,最大的头围不足43 cm。小头症患儿大脑发育障碍常常伴有不同程度的智力低下,有的患儿还会出现癫痫、运动障碍、语言障碍及其他行为异常[10]。造成小头症的原因有很多,大致可分成两类:一类是由遗传因素引起,即染色体或基因突变所导致;另一类是由环境因素引起,即胎儿在妊娠早期受到各种有害因素影响包括营养不良、中毒、物理或化学影响以及子宫感染(弓形虫病,风疹,疱疹,梅毒,巨细胞病毒及艾滋病毒)所导致。2015年,一种虫媒病毒(寨卡病毒)在美洲和热带地区爆发导致小头症新生儿的大量产生引发了全世界的关注[11]。通过动物模型验证,科学家已经证实寨卡病毒感染确实会导致小头症的发生[12~14]。
常染色体隐性遗传小头畸形(autosomal recessive primary microcephaly, MCPH)是一种比较少见的神经分裂异常引起的脑发育疾病。病人主要表现为头围减小并伴有不同程度的智力衰退[8]。目前发现的与MCPH有关的25个基因包括:()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()、()和()[15]。全世界范围内,超过50%的MCPH患者是由于()和)基因突变造成的[8]。本文通过查询小鼠基因组信息(Mouse Genome Informatics, MGI)和美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)数据库对已命名的25个MCPH相关蛋白的表达模式、细胞定位、分子生物学功能、表型及动物模型等进行了综述,旨在提升人们对MCPH发病机制的认知并加深人们对神经元生成及脑尺寸大小调控机理的理解。
1 MCPH基因的时空特异性表达
结合MGI和NCBI网站数据分析发现,相关基因存在非常明显的时空特异性表达(图1,图2)。MCPH作为一类神经发育疾病相关蛋白,大多数与细胞分裂相关,因此在细胞分裂活跃的组织表达较高。MGI数据库(基于免疫组化和RNA原位杂交结果)结果显示,有21个基因都在神经系统表达,仅和未在神经系统中检测到表达。除神经系统外,大部分在视觉系统、生殖系统和消化系统中也有表达。NCBI数据(基于正常组织RNAseq)显示在人体组织中,已发现的25个中有11个基因在睾丸(相对其他组织)中表达量最高,分别是()、()、()、()、(STIL)、(Cep152)、()、(MFSD2A)、(ANKLE2)、()和()。另外,还有4个基因在睾丸(相对其他组织)中表达量次高,其中包括()、(SAS-6)、(CIT)和()。主要原因可能是基因在分裂旺盛的组织表达较高,而睾丸在产生精子过程中首先会产生很多精原干细胞。

图1 Mcph在小鼠不同组织的表达模式
根据小鼠基因组信息(Mouse Genome Informatics, MGI)数据库总结得到22个基因在不同组织的表达模式。
此外,通过分析NCBI数据库RNA-seq结果,将25个基因在小鼠脑发育不同阶段的表达情况进行汇总,结果发现大部分在脑发育早期(胚胎11.5天)表达量较高,随着发育的进行(胚胎18天)表达量逐渐下降,在成年脑皮质中表达量更低甚至几乎检测不到(图2)。基因的时空特异性表达决定其在调控脑发育和育性方面起着非常重要的作用。
2 MCPH蛋白在细胞中的定位
通过蛋白序列分析或结构预测发现,大部分MCPH蛋白定位在细胞骨架和细胞核中(图3)。中心体是微管组织中心,大部分MCPH蛋白都定位在有丝分裂装置如中心体或纺锤体上[16]。在脑发育过程中,中心体和纺锤体的正确组装对于产生和维持正常的神经细胞的数量起着非常关键的作用。MCPH蛋白缺失或突变会干扰中心体或纺锤体的正常形成,影响细胞周期及DNA复制等过程,进而影响神经前体细胞的增殖、分化和凋亡等过程,最终导致神经元数量减少并形成偏小的大脑[17~20]。MCPH2/WDR62表现出非常明显的细胞周期依赖性表达。在有丝分裂中期或前中期,WDR62主要集中在纺锤体极点,而在有丝分裂间期WDR62则弥散地分布在细胞质中[21]。在细胞有丝分裂间期,高尔基体是除中心体外的另一个微管组织中心。有些MCPH蛋白定位在高尔基体上,如MCPH3 (CDK5RAP2)通过ATP及中心体依赖的形式定位在高尔基体上[22]。MCPH19 (COPB2,β-辅酶亚基)以GTP依赖形式结合在高尔基体膜上并对高尔基体的形成及囊泡运输过程起重要作用[23]。还有些MCPH蛋白(MCPH15/MFSD2A和MCPH16/ANKLE2)则定位在内质网上[24,25](图3)。

图2 Mcph基因在小鼠脑发育不同阶段的表达情况
根据美国国立生物技术信息中心(National Center for Biotechnology Information, NCBI)数据库总结得到不同基因在小鼠不同发育阶段中枢神经系统的表达情况。蓝色不同强度代表表达强弱,越接近蓝色表达越强,越接近白色表达越弱。
3 MCPH蛋白参与的生物学过程
动物个体发育是一个复杂而精细的过程。大部分MCPH蛋白在发育过程参与细胞成分组装进而调控细胞的增殖分化及系统发育等过程(图4)。大脑的大小是通过调节神经干细胞增殖、分化和凋亡的平衡来控制的[26]。目前研究得比较多的一些MCPH蛋白(如MCPH5/ASPM、MCPH2/WDR62和MCPH6/ CENPJ等)在神经干细胞的增殖或分化过程中起着重要作用[20,27~30]。神经迁移与神经发生和大脑尺寸调控密切相关。在大脑皮层形成过程中,正常的神经迁移是至关重要的一个环节,是构成大脑皮层复杂的组织结构及特殊回路的前提和基础。一些MCPH蛋白(如MCPH5和MCPH6)参与调控大脑皮层神经细胞的迁移[27,28]。一些MCPH蛋白(如MCPH1)则参与调控细胞凋亡或DNA损伤过程[31]。一些MCPH蛋白(如MCPH2/WDR62、MCPH3/ CDK5RAP2和MCPH5/ASPM)参与调控微管的组装、聚合或解聚[32~34]。MCPH7/STIL参与小鼠胚胎体轴的特化和神经管的发育[35]。MCPH18/WDFY3通过调控自噬过程来影响大脑发育[36]。
4 MCPH突变表型
4.1 MCPH突变病人的表型
MCPH病人一般比较矮小,身高和体重亦低于正常值下限。MCPH是一种神经发育障碍疾病,并且大部分基因在神经系统高表达,因此基因突变病人的表型主要集中在神经系统(图5)。大脑体积减小和皮质发育不良是MCPH病人的主要特征。同时一些MCPH病人还表现出小脑、脑干及胼胝体发育不良[15]。MCPH病人在行为上主要表现为不同程度的智力障碍,部分表现出运动及语言障碍,也有部分病人表现出共济失调、癫痫及先天性耳聋等症状[37~39]。
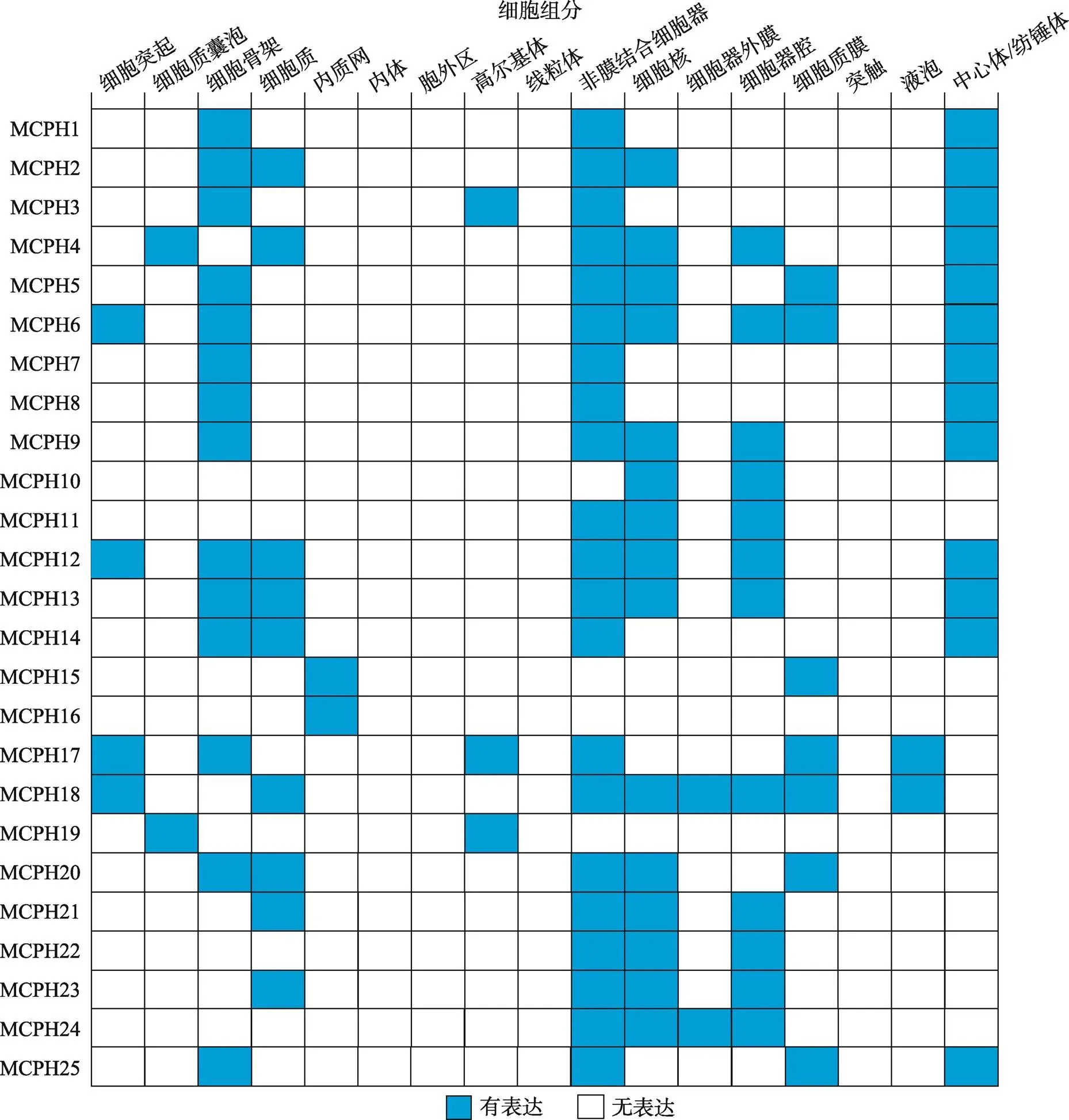
图3 MCPH蛋白的细胞定位
根据小鼠基因组信息(Mouse Genome Informatics, MGI)数据库总结得到25个MCPH蛋白在不同细胞组分中的定位。
有丝分裂过程和分离缺陷、微管和纺锤体异常以及DNA损伤和细胞周期异常等过程在病人来源或基因突变的细胞中被发现[21,40~44]。也有研究表明由于神经前体细胞中RNA加工模式的不同是导致MCPH突变影响神经系统的原因[45]。根据MGI数据库显示大部分突变会影响有丝分裂引起不同组织的多种表型,同时也会影响减数分裂引起生殖系统出现问题(图5)。
4.2 MCPH相关动物模型的构建
研究MCPH蛋白功能的动物模型主要有斑马鱼()、果蝇()和小鼠()。MCPH除在神经系统发育上表现出小头症的表型外,在个体生长发育过程中也起着重要作用(表1)。另外,MCPH蛋白在生殖系统中高表达,因此,MCPH蛋白缺失会导致小鼠生殖细胞发育缺陷进而引起育性降低,甚至不育[46~49]。近年来,有研究表明缺失也会影响小鼠听力并引发中耳炎[50]。由于MCPH蛋白对动物生长发育的关键作用,一些MCPH蛋白缺失甚至会导致动物胚胎致死或细胞凋亡。基因敲除小鼠的构建为表型分析和MCPH致病机制的研究提供了有效的动物模型。但由于小鼠大脑与人脑相比没有脑回结构及更多类型的神经前体细胞,基因敲除小鼠有时不能很好地模拟小头症的表型。2016年,科学家利用TALEN技术制备了突变体食蟹猴(),等位基因突变食蟹猴表现出头围减小、胼胝体发育不良以及上肢痉挛等特征模拟了大部分小头症病人的临床表型[51]。近年来有研究在雪貂()中敲除,敲除雪貂表现出更为明显的小头症表型[52]。
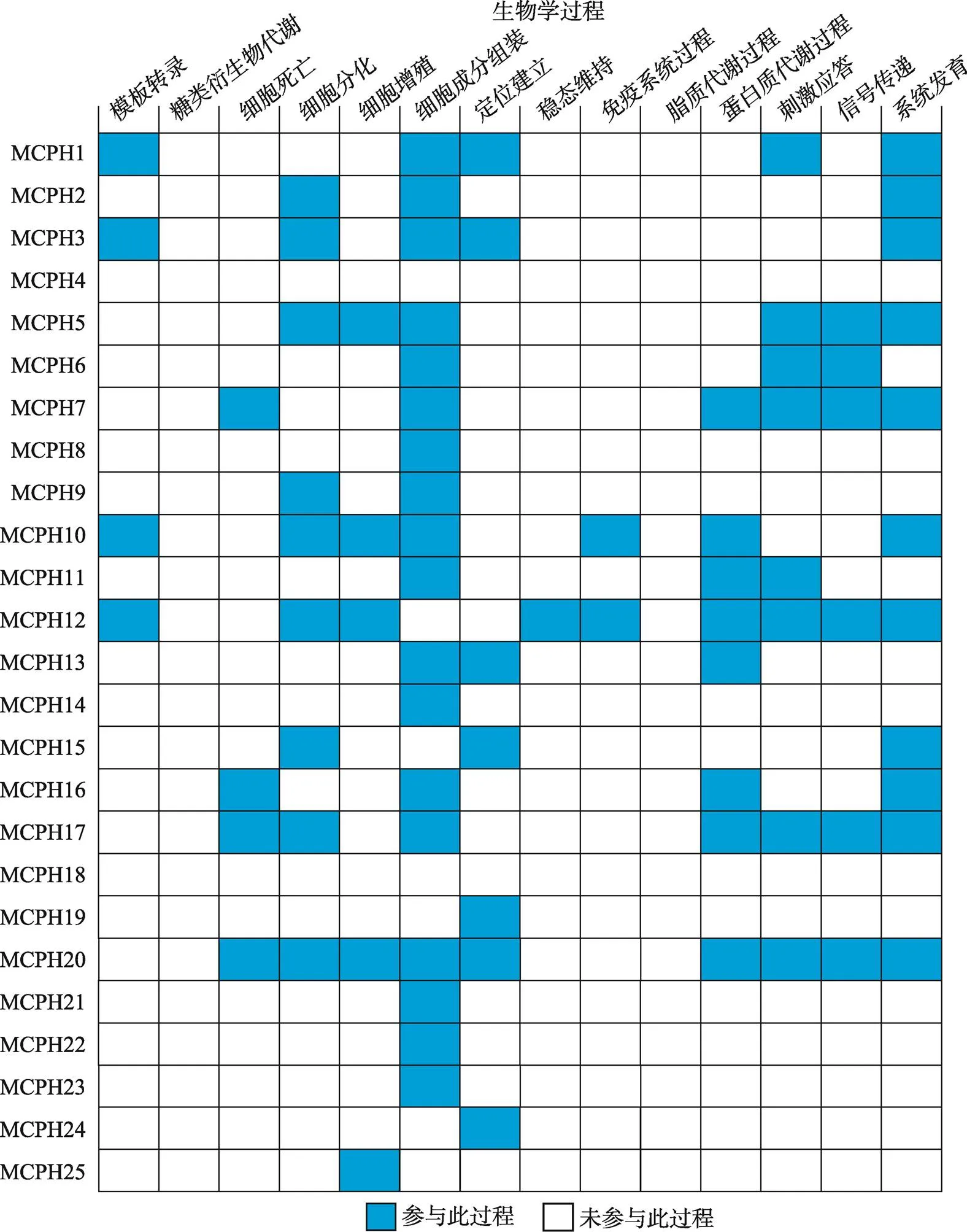
图4 MCPH蛋白参与的生物学过程
根据小鼠基因组信息(Mouse Genome Informatics, MGI)数据库总结得到25个MCPH蛋白参与的生物学过程。

图5 Mcph基因突变相关表型
根据小鼠基因组信息(Mouse Genome Informatics, MGI)数据库总结得到25个基因突变的表型。
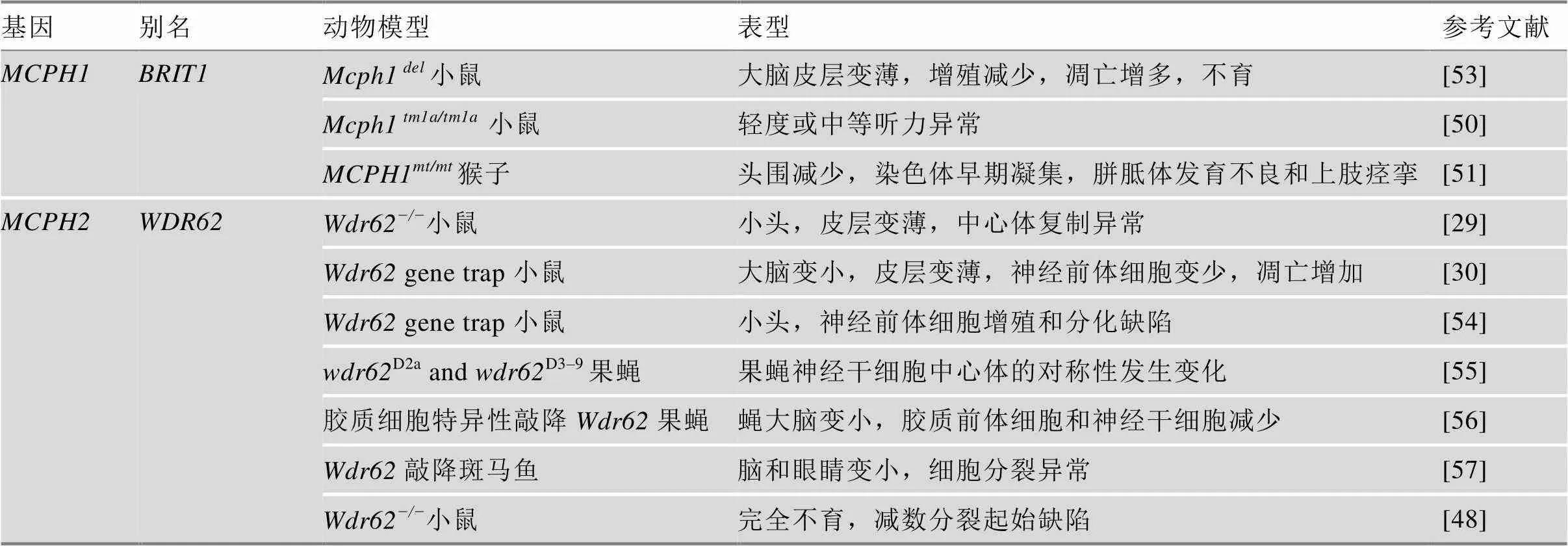
表1 MCPH相关动物模型
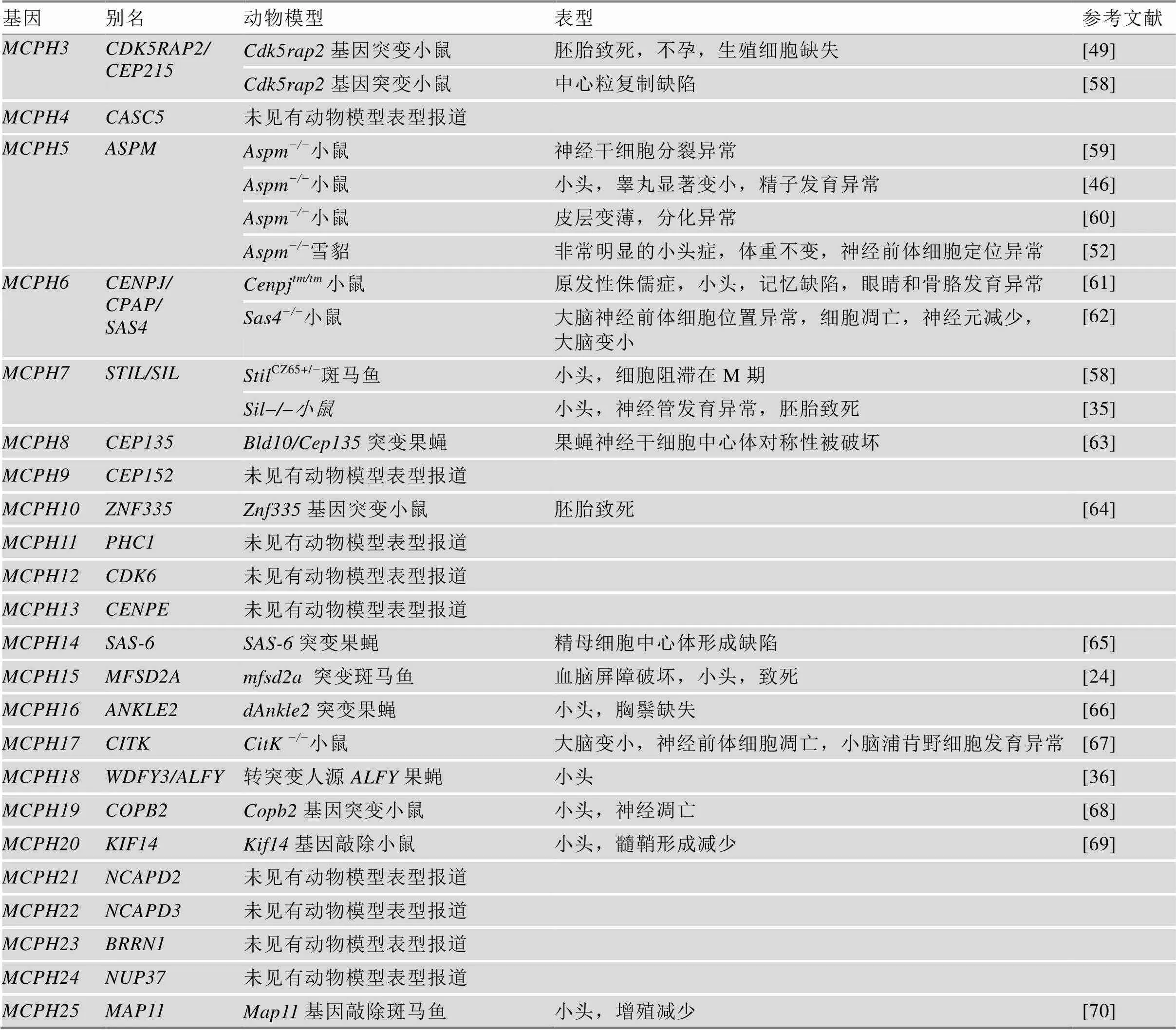
续表
5 MCPH分子机制研究
5.1 MCPH蛋白功能预测
MCPH蛋白分子的功能是多样化的。根据MGI网站生物信息学分析预测一些MCPH蛋白如MCPH3、MCPH6、MCPH13、MCPH20和MCPH25主要参与结合细胞骨架蛋白。一些MCPH蛋白结合DNA (MCPH3、MCPH10和MCPH11)和糖类及其衍生物(MCPH12、MCPH13、MCPH17和MCPH20);还有一些蛋白则作为各种酶如水解酶(MCPH13和MCPH20)、转移酶(MCPH6、MCPH12、MCPH17和MCPH18)催化各种生物化学反应来调控生物体内的新陈代谢和能量转换(图6)。
5.2 MCPH参与的信号通路
MCPH蛋白通常调控细胞周期,因此很多MCPH蛋白都参与细胞周期相关信号通路的调控。MCPH1通过Chk1–Cdc25–Cdk1调控中心体及纺锤体的形成进而调控神经前体细胞的分裂[53]。MCPH1/ BRIT1还可以与E2F1结合调控CHK1和BRCA1参与DNA修复和细胞凋亡调控[71]。MCPH2/WDR62通过JNK信号通路调控神经前体细胞的增殖和分化[20]。同时,MEKK3和FBW7双向调控WDR62蛋白的稳定性进而调控大脑皮层神经干细胞稳态平衡[72]。MCPH3/CDK5RAP2与EB1结合调控微管的动态组装[32]。MCPH5/ASPM通过Wnt信号通路调控神经前体细胞的增殖和分化[27]。MCPH6/CPAP在大脑发育过程中作用于Ascl1下游来调控神经前体细胞的分裂及神经迁移[28]。MCPH7通过调控Sonic hedgehog (Shh)信号通路来调控胚胎体轴的发育[36]。MCPH9/CEP152与PLK4结合调控中心粒的复制[52]。MCPH9/CEP152和MCPH17/CITK被报道与p53信号通路相关[47,67]。MCPH18/ALFY通过调控自噬来调控经典Wnt信号通路[35]。MCPH19/COPB2通过上调YAP表达来调控细胞增殖[73]。
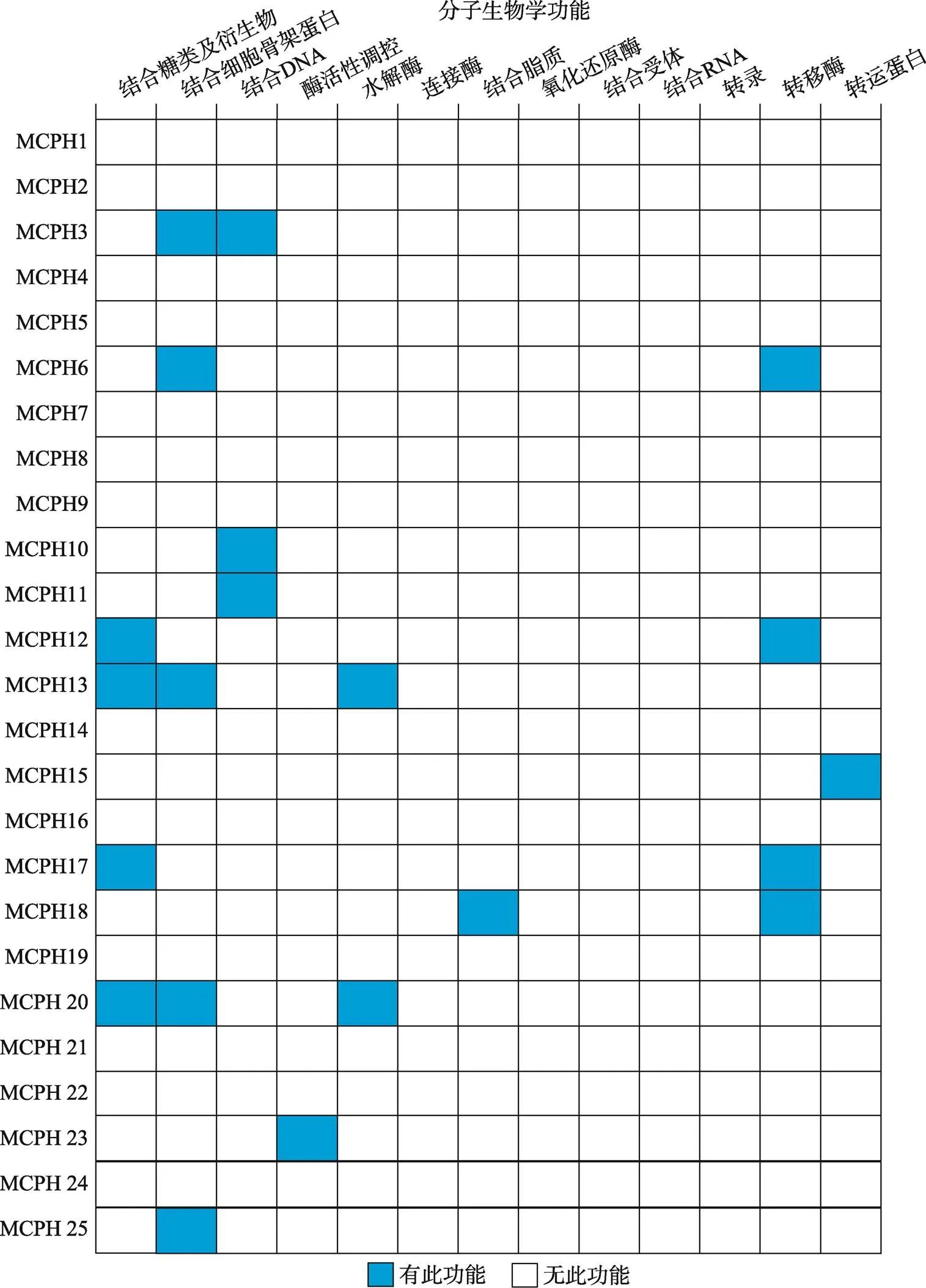
图6 MCPH蛋白的分子生物学功能
根据小鼠基因组信息(Mouse Genome Informatics, MGI)数据库总结得到25个MCPH蛋白的分子生物学功能。
6 结语与展望
现代人大脑的尺寸大小约是3百万年前人类祖先南方古猿人大脑的3倍,特别是大脑皮层及脑回数量增加达100倍[74]。大脑的尺寸大小及结构复杂度的增加必然伴随认知功能的增加[75]。由于大脑的尺寸大小及脑回数量与神经元的数量密切相关,因此研究那些影响神经细胞增殖、分化及凋亡的基因有利于了解脑发育过程及人类的进化过程。MCPH是一种常染色体隐性遗传小头畸形病症。本文对已报道的25个MCPH蛋白的表达、定位和功能进行了总结和概述,为研究大脑发育相关蛋白特别是MCPH蛋白的致病机制提供了理论依据和线索。在已报道的25个MCPH中,和突变导致的MCPH病人较多,因此相关研究也较多。同时根据被发现的顺序,一些较早发现的MCPH蛋白(MCPH1~MCPH10)的动物模型建立较多,而较晚发现的MCPH11~MCPH25只有少部分蛋白的动物模型已经建立。在已建立的动物模型中,大部分MCPH缺失或突变模型都会导致小头症的典型特征即大脑明显变小,还有一部分动物模型影响了生殖系统。导致小头症的分子机制研究主要集中在调控中心体或纺锤体的形成进而影响神经前体细胞的增殖分化和细胞凋亡。也有一些MCPH突变小鼠表现出小脑发育不良或神经元轴突发育异常。近年来,大脑类器官的建立为研究大脑疾病包括小头症的发病机制提供了很好的模型[76,77]。Gabriel等[77,78]利用人iPSC诱导的类脑模型研究MCPH6/CPAP的致病机制。鉴于大脑类器官模型在体外培养的局限性,构建小头症相关蛋白缺失或突变的动物模型仍是未来研究小头症发病机制的有效手段。
大脑的尺寸在一定程度上反应脑重量和脑容量的大小[79]。但是拥有一个更大的大脑并不意味着人类或动物在认知方面就表现得更好。病人的头围相对于其同年龄与性别的平均值明显大2个标准差以上会导致另外一种脑发育疾病称为巨脑症(macrocephaly)[80]。巨脑症主要是由于神经增殖和迁移异常引起,病人表现出智力低下并有部分伴有自闭症样行为[80~83]。因此,维持大脑尺寸的正常发育对于脑功能的正常发挥起着非常重要的作用。
[1] Thornton GK, Woods CG. Primary microcephaly: do all roads lead to Rome?, 2009, 25(11): 501– 510.
[2] Kang EC, Burdick KE, Kim JY, Duan X, Guo JU, Sailor KA, Jung DE, Ganesan S, Choi S, Pradhan D, Lu B, Avramopoulos D, Christian K, Malhotra AK, Song HJ, Ming GL. Interaction between FEZ1 and DISC1 in regulation of neuronal development and risk for schizophrenia., 2011, 72(4): 559–571.
[3] Kaindl AM, Passemard S, Kumar P, Kraemer N, Issa L, Zwirner A, Gerard B, Verloes A, Mani S, Gressens P. Many roads lead to primary autosomal recessive microcephaly., 2010, 90(3): 363–383.
[4] Gleeson JG. Neuronal migration disorders., 2001, 7(3): 167–171.
[5] Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, Chen Z, Zhang C, Christian KM, Song H, Ming GL, Xu Z. Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice., 2016, 7.
[6] Kim JY, Liu CY, Zhang FY, Duan X, Wen ZX, Song J, Feighery E, Lu B, Rujescu D, St Clair D, Christian K, Callicott JH, Weinberger DR, Song HJ, Ming GL. Interplay between DISC1 and GABA signaling regulates neurogenesis in Mice and Risk for schizophrenia., 2012, 148(5): 1051–1064.
[7] Dang T, Duan WY, Yu B, Tong DL, Cheng C, Zhang YF, Wu W, Ye K, Zhang WX, Wu M, Wu BB, An Y, Qiu ZL, Wu BL. Autism-associated dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development., 2018, 23(3): 747–758.
[8] Mahmood S, Ahmad W, Hassan MJ. Autosomal recessive primary microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum., 2011, 6: 39.
[9] Woods CG, Parker A. Investigating microcephaly., 2013, 98(9): 707–713.
[10] McDonell LM, Warman Chardon J, Schwartzentruber J, Foster D, Beaulieu CL, FORGE Canada Consortium, Majewski J, Bulman DE, Boycott KM. The utility of exome sequencing for genetic diagnosis in a familial microcephaly epilepsy syndrome., 2014, 14: 22.
[11] Kruger RP. Zika virus on the move., 2016, 164(4): 585–587.
[12] Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, Mysorekar IU, Diamond MS. Zika virus infection during pregnancy in mice causes placental damage and fetal demise., 2016, 165(5): 1081–1091.
[13] Li C, Xu D, Ye Q, Hong S, Jiang YS, Liu X, Zhang N, Shi L, Qin CF, Xu Z. Zika virus disrupts neural progenitor development and leads to microcephaly in mice., 2016, 19(5):120–126.
[14] Cugola FR, Fernandes IR, Russo FB, Freitas BC, Dias JLM, Guimarães KP, Benazzato C, Almeida N, Pignatari GC, Romero S, Polonio CM, Cunha I, Freitas CL, Brandão WN, Rossato C, Andrade DG, Faria Dde P, Garcez AT, Buchpigel CA, Braconi CT, Mendes E, Sall AA, Zanotto PM, Peron JP, Muotri AR, Beltrão-Braga PC. The brazilian zika virus strain causes birth defects in experimental models., 2016, 534(7606): 267–271.
[15] Zaqout S, Morris-Rosendahl D, Kaindl AM. Autosomal recessive primary microcephaly (MCPH): an update., 2017, 48(3): 135–142.
[16] Megraw TL, Sharkey JT, Nowakowski RS. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes., 2011, 21(8): 470–480.
[17] Manzini MC, Walsh CA. What disorders of cortical development tell us about the cortex: one plus one does not always make two., 2011, 21(3): 333–339.
[18] Cox J, Jackson AP, Bond J, Woods CG. What primary microcephaly can tell us about brain growth., 2006, 12(8): 358–366.
[19] Farag HG, Froehler S, Oexle K, Ravindran E, Schindler D, Staab T, Huebner A, Kraemer N, Chen W, Kaindl AM. Abnormal centrosome and spindle morphology in a patient with autosomal recessive primary microcephaly type 2 due to compound heterozygous, 2013, 8: 178.
[20] Xu D, Zhang F, Wang Y, Sun Y, Xu Z. Microcephaly- associated protein WDR62 regulates neurogenesis through JNK1 in the developing neocortex., 2014, 6(1): 104–116.
[21] Nicholas AK, Khurshid M, Désir J, Carvalho OP, Cox JJ, Thornton G, Kausar R, Ansar M, Ahmad W, Verloes A, Passemard S, Misson JP, Lindsay S, Gergely F, Dobyns WB, Roberts E, Abramowicz M, Woods CG. WDR62 is associated with the spindle pole and is mutated in human microcephaly., 2010, 42(11): 1010–1014.
[22] Wang Z, Wu T, Shi L, Zhang L, Zheng W, Qu JY, Niu R, Qi RZ. Conserved motif of CDK5RAP2 mediates its localization to centrosomes and the Golgi complex., 2010, 285(29): 22658–22665.
[23] Guo Y, Punj V, Sengupta D, Linstedt AD. Coat-tether interaction in golgi organization., 2008, 19(7): 2830–2843.
[24] Guemez-Gamboa A, Nguyen LN, Yang H, Zaki MS, Kara M, Ben-Omran T, Akizu N, Rosti RO, Rosti B, Scott E, Schroth J, Copeland B, Vaux KK, Cazenave-Gassiot A, Quek DQ, Wong BH, Tan BC, Wenk MR, Gunel M, Gabriel S, Chi NC, Silver DL, Gleeson JG. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome., 2015, 47(7): 809–813.
[25] Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium., 2011, 12(5): 449–462.
[26] Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings., 2005, 76(5): 717–728.
[27] Buchman JJ, Durak O, Tsai LH. ASPM regulates Wnt signaling pathway activity in the developing brain., 2011, 25(18): 1909–1914.
[28] Garcez PP, Diaz-Alonso J, Crespo-Enriquez I, Castro D, Bell D, Guillemot F. Cenpj/CPAP regulates progenitor divisions and neuronal migration in the cerebral cortex downstream of ascl1., 2015, 6: 6474.
[29] Jayaraman D, Kodani A, Gonzalez DM, Mancias JD, Mochida GH, Vagnoni C, Johnson J, Krogan N, Harper JW, Reiter JF, Yu TW, Bae BI, Walsh CA. Microcephaly proteins Wdr62 and aspm define a mother centriole complex regulating centriole biogenesis, apical complex, and cell fate., 2016, 92(4): 813–828.
[30] Chen JF, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L. Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size., 2014, 5: 3885.
[31] Zhou ZW, Tapias A, Bruhn C, Gruber R, Sukchev M, Wang ZQ. DNA damage response in microcephaly development of MCPH1 mouse model., 2013, 12(8): 645–655.
[32] Fong KW, Hau SY, Kho YS, Jia Y, He L, Qi RZ. Interaction of CDK5RAP2 with EB1 to track growing microtubule tips and to regulate microtubule dynamics., 2009, 20(16): 3660–3670.
[33] Jiang K, Rezabkova L, Hua SS, Liu Q, Capitani G, Altelaar AFM, Heck AJR, Kammerer RA, Steinmetz MO, Akhmanova A. Microtubule minus-end regulation at spindle poles by an ASPM-katanin complex., 2017, 19(5): 480–492.
[34] Lim NR, Yeap YY, Zhao TT, Yip YY, Wong SC, Xu D, Ang CS, Williamson NA, Xu Z, Bogoyevitch MA, Ng DC. Opposing roles for JNK and aurora a in regulating the association of WDR62 with spindle microtubules., 2015, 128(3): 527–540.
[35] Izraeli S, Lowe LA, Bertness VL, Good DJ, Dorward DW, Kirsch IR, Kuehn MR. The SIL gene is required for mouse embryonic axial development and left-right specification., 1999, 399(6737): 691–694.
[36] Kadir R, Harel T, Markus B, Perez Y, Bakhrat A, Cohen I, Volodarsky M, Feintsein-Linial M, Chervinski E, Zlotogora J, Sivan S, Birnbaum RY, Abdu U, Shalev S, Birk OS. ALFY-Controlled DVL3 autophagy regulates Wnt signaling, determining human brain size., 2016, 12(3): e1005919.
[37] Abdullah U, Farooq M, Mang Y, Marriam Bakhtiar S, Fatima A, Hansen L, Kjaer KW, Larsen LA, Faryal S, Tommerup N, Mahmood Baig S. A novel mutation in CDK5RAP2 gene causes primary microcephaly with speech impairment and sparse eyebrows in a consanguineous pakistani family., 2017, 60(12): 627–630.
[38] Darvish H, Esmaeeli-Nieh S, Monajemi GB, Mohseni M, Ghasemi-Firouzabadi S, Abedini SS, Bahman I, Jamali P, Azimi S, Mojahedi F, Dehghan A, Shafeghati Y, Jankhah A, Falah M, Soltani Banavandi MJ, Ghani M, Garshasbi M, Rakhshani F, Naghavi A, Tzschach A, Neitzel H, Ropers HH, Kuss AW, Behjati F, Kahrizi K, Najmabadi H. A clinical and molecular genetic study of 112 Iranian families with primary microcephaly., 2010, 47(12): 823–828.
[39] Shen J, Eyaid W, Mochida GH, Al-Moayyad F, Bodell A, Woods CG, Walsh CA. ASPM mutations identified in patients with primary microcephaly and seizures., 2005, 42(9): 725–729.
[40] Miyamoto T, Akutsu SN, Fukumitsu A, Morino H, Masatsuna Y, Hosoba K, Kawakami H, Yamamoto T, Shimizu K, Ohashi H, Matsuura S. PLK1-mediated phosphorylation of WDR62/MCPH2 ensures proper mitotic spindle orientation., 2017, 26(22): 4429–4440.
[41] Arroyo M, Kuriyama R, Trimborn M, Keifenheim D, Cañuelo A, Sánchez A, Clarke DJ, Marchal JA. MCPH1, mutated in primary microcephaly, is required for efficient chromosome alignment during mitosis., 2017, 7(1): 13019.
[42] Issa L, Mueller K, Seufert K, Kraemer N, Rosenkotter H, Ninnemann O, Buob M, Kaindl AM, Morris-Rosendahl DJ. Clinical and cellular features in patients with primary autosomal recessive microcephaly and a novel CDK5RAP2 mutation., 2013, 8: 59.
[43] Guernsey DL, Jiang HY, Hussin J, Arnold M, Bouyakdan K, Perry S, Babineau-Sturk T, Beis J, Dumas N, Evans SC, Ferguson M, Matsuoka M, Macgillivray C, Nightingale M, Patry L, Rideout AL, Thomas A, Orr A, Hoffmann I, Michaud JL, Awadalla P, Meek DC, Ludman M, Samuels ME. Mutations in centrosomal protein CEP152 in primary microcephaly families linked to MCPH4., 2010, 87(1): 40–51.
[44] Hussain MS, Baig SM, Neumann S, Nürnberg G, Farooq M, Ahmad I, Alef T, Hennies HC, Technau M, Altmüller J, Frommolt P, Thiele H, Noegel AA, Nürnberg P. A truncating mutation of CEP135 causes primary microcephaly and disturbed centrosomal function., 2012, 90(5): 871–878.
[45] Omer Javed A, Li Y, Muffat J, Su KC, Cohen MA, Lungjangwa T, Aubourg P, Cheeseman IM, Jaenisch R. Microcephaly modeling of kinetochore mutation reveals a brain-specific phenotype., 2018, 25(2): 368– 382.e5.
[46] Pulvers JN, Bryk J, Fish JL, Wilsch-Bräuninger M, Arai Y, Schreier D, Naumann R, Helppi J, Habermann B, Vogt J, Nitsch R, Tóth A, Enard W, Pääbo S, Huttner WB. Mutations in mouse Aspm (abnormal spindle-like microcephaly associated) cause not only microcephaly but also major defects in the germline., 2010, 107(38): 16595–16600.
[47] Marjanović M, Sánchez-Huertas C, TerréB, Gómez R, Scheel JF, Pacheco S, Knobel PA, Martínez-Marchal A, Aivio S, Palenzuela L, Wolfrum U, McKinnon PJ, Suja JA, Roig I, Costanzo V, Lüders J, Stracker TH. CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination., 2015, 6: 7676.
[48] Zhou Y, Qin Y, Qin Y, Xu B, Guo T, Ke HN, Chen M, Zhang L, Han F, Li Y, Chen M, Behrens A, Wang Y, Xu Z, Chen ZJ, Gao F. Wdr62 is involved in female meiotic initiation via activating JNK signaling and associated with POI in humans., 2018, 14(8): e1007463.
[49] Zaqout S, Bessa P, Krämer N, Stoltenburg-Didinger G, Kaindl AM. CDK5RAP2 is required to maintain the germ cell pool during embryonic development., 2017, 8(2): 198–204.
[50] Chen J, Ingham N, Clare S, Raisen C, Vancollie VE, Ismail O, McIntyre RE, Tsang SH, Mahajan VB, Dougan G, Adams DJ, White JK, Steel KP. Mcph1-deficient mice reveal a role for MCPH1 in otitis media., 2013, 8(3): e58156.
[51] Ke Q, Li W, Lai X, Chen H, Huang L, Kang Z, Li K, Ren J, Lin X, Zheng H, Huang W, Ma Y, Xu D, Chen Z, Song X, Lin X, Zhuang M, Wang T, Zhuang F, Xi J, Mao FF, Xia H, Lahn BT, Zhou Q, Yang S, Xiang AP. TALEN-based generation of a cynomolgus monkey disease model for human microcephaly., 2016, 26(9): 1048–1061.
[52] Johnson MB, Sun X, Kodani A, Borges-Monroy R, Girskis KM, Ryu SC, Wang PP, Patel K, Gonzalez DM, Woo YM, Yan Z, Liang B, Smith RS, Chatterjee M, Coman D, Papademetris X, Staib LH, Hyder F, Mandeville JB, Grant PE, Im K, Kwak H, Engelhardt JF, Walsh CA, Bae BI. Aspm knockout ferret reveals an evolutionary mechanism governing cerebral cortical size., 2018, 556(7701): 370–375.
[53] Gruber R, Zhou ZW, Sukchev M, Joerss T, Frappart PO, Wang ZQ. MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1-Cdc25 pathway., 2011, 13(11): 1325–1334.
[54] Sgourdou P, Mishra-Gorur K, Saotome I, Henagariu O, Tuysuz B, Campos C, Ishigame K, Giannikou K, Quon JL, Sestan N, Caglayan AO, Gunel M, Louvi A. Disruptions in asymmetric centrosome inheritance and WDR62-Aurora kinase B interactions in primary microcephaly., 2017, 7: 43708.
[55] Ramdas Nair A, Singh P, G Salvador arcia D, Rodriguez- Crespo D, Egger B, Cabernard C. The Microcephaly- Associated protein Wdr62/CG7337 Is required to maintain centrosome asymmetry in drosophila neuroblasts., 2016, 14(5): 1100–1113.
[56] Lim NR, Shohayeb B, Zaytseva O, Mitchell N, Millard SS, Ng DCH, Quinn LM. Glial-Specific functions of microcephaly protein WDR62 and interaction with the mitotic kinase AURKA are essential for drosophila brain growth., 2017, 9(1): 32–41.
[57] Novorol C, Burkhardt J, Wood KJ, Iqbal A, Roque C, Coutts N, Almeida AD, He J, Wilkinson CJ, Harris WA. Microcephaly models in the developing zebrafish retinal neuroepithelium point to an underlying defect in metaphase progression., 2013, 3(10): 130065.
[58] Barrera JA, Kao LR, Hammer RE, Seemann J, Fuchs JL, Megraw TL. CDK5RAP2 regulates centriole engagement and cohesion in mice., 2010, 18(6): 913–926.
[59] Capecchi MR, Pozner A. ASPM regulates symmetric stem cell division by tuning cyclin E ubiquitination., 2015, 6: 8763.
[60] Fujimori A, Itoh K, Goto S, Hirakawa H, Wang B, Kokubo T, Kito S, Tsukamoto S, Fushiki S. Disruption of Aspm causes microcephaly with abnormal neuronal differentiation., 2014, 36(8): 661–669.
[61] McIntyre RE, Lakshminarasimhan Chavali P, Ismail O, Carragher DM, Sanchez-Andrade G, Forment JV, Fu B, Del Castillo Velasco-Herrera M, Edwards A, van der Weyden L, Yang F, Sanger Mouse Genetics Project, Ramirez-Solis R, Estabel J, Gallagher FA, Logan DW, Arends MJ, Tsang SH, Mahajan VB, Scudamore CL, White JK, Jackson SP, Gergely F, Adams DJ. Disruption of mouse cenpj, a regulator of centriole biogenesis, phenocopies seckel syndrome., 2012, 8(11): e1003022.
[62] Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH. Cortical neurogenesis in the absence of centrioles., 2014, 17(11): 1528–1535.
[63] Roque H, Wainman A, Richens J, Kozyrska K, Franz A, Raff JW.Cep135/Bld10 maintains proper centriole structure but is dispensable for cartwheel formation, 2012, 125(Pt 23): 5881–5886.
[64] Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, Tatarakis A, Mahajan MA, Samuels HH, Shi Y, Golden JA, Mahajnah M, Shenhav R, Walsh CA. Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation., 2012, 151(5): 1097–1112.
[65] Hilbert M, Noga A, Frey D, Hamel V, Guichard P, Kraatz SH, Pfreundschuh M, Hosner S, Flückiger I, Jaussi R, Wieser MM, Thieltges KM, Deupi X, Müller DJ, Kammerer RA, Gönczy P, Hirono M, Steinmetz MO. SAS-6 engineering reveals interdependence between cartwheel and microtubules in determining centriole architecture., 2016, 18(4): 393–403.
[66] Yamamoto S, Jaiswal M, Charng WL, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H, Haelterman NA, Xiong B, Zhang K, Bayat V, David G, Li T, Chen K, Gala U, Harel T, Pehlivan D, Penney S, Vissers LELM, de Ligt J, Jhangiani SN, Xie YJ, Tsang SH, Parman Y, Sivaci M, Battaloglu E, Muzny D, Wan YW, Liu Z, Lin-Moore AT, Clark RD, Curry CJ, Link N, Schulze KL, Boerwinkle E, Dobyns WB, Allikmets R, Gibbs RA, Chen R, Lupski JR, Wangler MF, Bellen HJ. A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases., 2014, 159(1): 200–214.
[67] Bianchi FT, Tocco C, Pallavicini G, Liu Y, VernìF, Merigliano C, Bonaccorsi S, El-Assawy N, Priano L, Gai M, Berto GE, Chiotto AM, SgròF, Caramello A, Tasca L, Ala U, Neri F, Oliviero S, Mauro A, Geley S, Gatti M, Di Cunto F. Citron kinase deficiency leads to chromosomal instability and TP53-Sensitive microcephaly., 2017, 18(7): 1674–1686.
[68] DiStasio A, Driver A, Sund K, Donlin M, Muraleedharan RM, Pooya S, Kline-Fath B, Kaufman KM, Prows CA, Schorry E, Dasgupta B, Stottmann RW. Copb2 is essential for embryogenesis and hypomorphic mutations cause human microcephaly., 2017, 26(24): 4836–4848.
[69] Fujikura K, Setsu T, Tanigaki K, Abe T, Kiyonari H, Terashima T, Sakisaka T. Kif14 mutation causes severe brain malformation and hypomyelination., 2013, 8(1): e53490.
[70] Perez Y, Bar-Yaacov R, Kadir R, Wormser O, Shelef I, Birk OS, Flusser H, Birnbaum RY. Mutations in the microtubule-associated protein MAP11 (C7orf43) cause microcephaly in humans and zebrafish., 2019, 142(3): 574–585.
[71] Yang SZ, Lin FT, Lin WC. MCPH1/BRIT1 cooperates with E2F1 in the activation of checkpoint, DNA repair and apoptosis., 2008, 9(9): 907–915.
[72] Xu D, Yao M, Wang Y, Yuan L, Hoeck JD, Yu J, Liu L, Yeap YYC, Zhang W, Zhang F, Feng Y, Ma T, Wang Y, Ng DCH, Niu X, Su B, Behrens A, Xu Z. MEKK3 coordinates with FBW7 to regulate WDR62 stability and neurogenesis., 2018, 16(12): e2006613.
[73] Luo X, Liu Y, Feng W, Lei L, Du Y, Wu J, Wang S. NUP37, a positive regulator of YAP/TEAD signaling, promotes the progression of hepatocellular carcinoma., 2017, 8(58): 98004–98013.
[74] Clark DA, Mitra PP, Wang SS. Scalable architecture in mammalian brains., 2001, 411(6834): 189–193.
[75] Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution., 1995, 18(9): 383–388.
[76] Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly., 2013, 501(7467): 373–379.
[77] Gabriel E, Wason A, Ramani A, Gooi LM, Keller P, Pozniakovsky A, Poser I, Noack F, Telugu NS, Calegari F, Šarić T, Hescheler J, Hyman AA, Gottardo M, Callaini G, Alkuraya FS, Gopalakrishnan J. CPAP promotes timely cilium disassembly to maintain neural progenitor pool., 2016, 35(8): 803–819.
[78] Gabriel E, Gopalakrishnan J. Generation of iPSC-derived human brain organoids to model early neurodevelopmental disorders., 2017, (122).
[79] Bartholomeusz HH, Courchesne E, Karns CM. Relationship between head circumference and brain volume in healthy normal toddlers, children, and adults., 2002, 33(5): 239–241.
[80] Pavone P, Praticò AD, Rizzo R, Corsello G, Ruggieri M, Parano E, Falsaperla R. A clinical review on megalencephaly: a large brain as a possible sign of cerebral impairment., 2017, 96(26): e6814.
[81] Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism., 2003, 290(3): 337–344.
[82] Courchesne E. Brain development in autism: early overgrowth followed by premature arrest of growth., 2004, 10(2): 106–111.
[83] Klein S, Sharifi-Hannauer P, Martinez-Agosto JA. Macrocephaly as a clinical indicator of genetic subtypes in autism., 2013, 6(1): 51–56.
Update on autosomal recessive primary microcephaly (MCPH)-associated proteins
Yujie Wang, Xiaokun Zhou, Dan Xu
Brain development diseases refer to a group of diseases that affect the development of the brain or the central nervous system. Autosomal recessive primary microcephaly (MCPH) is a typical neurodevelopmental disorder characterized by a decreased brain size, mental retardation and abnormal behaviors. To date, at least 25 genes have been discovered to cause MCPH when mutated. These genes were namedaccording to the discovery order. MCPH proteins play important roles in regulating brain developmental signaling pathways. Here, we provide a timely review of the expression patterns, cellular localization, molecular functions, phenotypes, as well as animal models of these 25 MCPH proteins that will expedite our understanding of the pathogenesis of brain disorders at both molecular and cellular levels.
microcephaly; expression pattern; cellular component; phenotype overview; molecular function; animal models
2019-04-18;
2019-05-15
福建省自然科学基金项目(编号:2018J01730)资助[Supported by the Natural Science Foundation of the Fujian Province (No. 2018J01730)]
王玉杰,硕士研究生,专业方向:细胞生物学。E-mail: yjwang9412@163.com
徐丹,博士,助理研究员,研究方向:脑发育相关疾病。E-mail: xu200828@163.com
10.16288/j.yczz.19-070
2019/6/11 17:00:20
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20190611.1700.001.html
(责任编委: 许执恒)
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
世界科学技术-中医药现代化(2021年5期)2021-11-05
世界科学技术-中医药现代化(2020年2期)2020-07-25
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
现代园艺(2017年21期)2018-01-03
作文教学研究(2016年1期)2016-07-05
中国中医眼科杂志(2015年1期)2015-12-28
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年8期)2015-06-22
