超高效液相色谱-串联质谱法测定人血浆中苯海索
2019-11-07 01:59:30杨雨菲夏云燕邹巧根
色谱 2019年12期
杨雨菲, 夏云燕, 吴 莎, 邹巧根*
(1.南京工业大学生物与制药工程学院, 江苏 南京 211816; 2.南京工业大学药学院, 江苏 南京 211816)
苯海索(C20H31NO, trihexyphenidyl),化学名为(±)-α-环己基-α-苯基-1-哌啶丙醇。苯海索为抗胆碱能药物,通过抑制黑质-纹状体内胆碱能神经活性抑制副交感神经,用于缓解运动障碍、震颤等症状。苯海索通常被用于帕金森病的治疗,临床研究[1,2]显示,单独使用苯海索能够显著降低帕金森症状的发生,并且不良反应相对较少。苯海索还可用于精神疾病的治疗及辅助治疗[3],可用于减缓精神分裂症阴性症状、精神障碍、静坐不能、流涎和撤药等症状。有临床研究[4]将苯海索与抗精神病药物(包括氯氮平、维思通、奋乃静、喹硫平、氯丙嗪和氟哌啶醇)联用,用于减轻或预防由抗精神病药物引起的锥体外系不良反应。苯海索还可用于治疗脑性瘫痪肌张力障碍[5],疗效良好的同时无严重不良反应。
基于苯海索熔沸点较低的性质,早期对于苯海索的体内检测多利用气相色谱偶联不同检测器进行定量分析[6],也有通过液相色谱-质谱法[7]和放射性免疫分析法[8]对苯海索进行定量检测。目前对于苯海索血浆样品的前处理方法主要采用液液萃取法和固相萃取法。液液萃取法常用的萃取剂为乙酸乙酯[7]、乙酸乙酯-正己烷(1∶3, v/v)[9]、环己烷[10]、庚烷[11]和苯-乙酸乙酯(1∶1, v/v)[12]等。固相萃取法利用Sep-Pak C18固相萃取柱[11]、Isolute HCX-5 (100 mg)混合模式固相萃取柱[13]提取样品。有报道表明,生物样本分析采用区别于传统前处理方法的手段,包括利用新体系对样品进行萃取[14]、衍生化前处理[15]等。针对苯海索体内研究的新方向包括利用新型电极碳糊电极(CPE)对苯海索片剂及尿液样本进行检测[16]。此外,利用铈(IV)(Ce(IV))或高锰酸钾(KMnO4)在酸性介质中诱导铜纳米团簇(CuNCs)化学发光(CL)的方法也被用于苯海索的定量检测[17]。通过对血浆[7-11,17]、血清[18]、全血[6,12]、头发[19]和尿液样本[10,13,16]的检测,可对苯海索进行临床用药监测和药代动力学研究等。
现有的对于苯海索血浆样品的检测方法仍存在前处理方法繁琐耗时、检测分析时间长、不适用于大批量样品检测等问题,因而有必要开发一种简便快捷、灵敏、适用于高通量分析的方法,以满足人血浆中苯海索定量分析的检测要求。
本文建立了一种超高效液相色谱-串联质谱(UPLC-MS/MS)测定人血浆中苯海索含量的方法。采用以甲醇为沉淀剂的蛋白质沉淀前处理方法,提取过程快速简便且回收率高,灵敏度良好。应用本方法考察了空腹及餐后健康受试者的药代动力学行为,并进行了生物等效性评价。
1 实验部分
1.1 仪器、试剂与材料
超高效液相色谱仪配有LC-30AD液相色谱泵、DGU-20A5R脱气机、CTO-20AC柱温箱和SIL-30ACMP自动进样器(Shimadzu公司,日本); API5500型三重四极杆质谱仪、Turbo Spray离子源(Applied Biosytems/Sciex公司,美国); XP6微量天平(Mettler Toledo公司,瑞士); Heraeus Muitifuge X1R离心机(Thermo Fisher公司,美国); Milli DirectQ纯水仪(Millipore公司,美国)。
盐酸苯海索对照品(纯度100%,批号:1.2)购自法国European Pharmacopoeia公司;盐酸苯海索-d11(纯度98.0%,批号:13-YSW-162-1)购自加拿大TRC公司。盐酸苯海索片受试制剂(2 mg,批号:1806001)购自天津力生制药有限公司;盐酸苯海索片参比制剂(2 mg,批号:1257989M)购自美国Watson公司。乙腈和甲醇购自德国Merck公司;甲酸和醋酸铵购自阿拉丁公司;异丙醇购自永华化学科技江苏有限公司,以上试剂均为色谱级。超纯水为实验室纯水仪自制。
1.2 标准溶液的配制
称取适量盐酸苯海索对照品,用甲醇溶解,配制成质量浓度为1.00 mg/mL的标准储备液,于2~8 ℃保存;用乙腈-水(30∶70, v/v)配制质量浓度为2.00、4.00、20.0、40.0、100、300、700和800 ng/mL的标准工作溶液和质量浓度为2.00、5.00、40.0和600 ng/mL的质控样品工作溶液。
称取适量盐酸苯海索-d11对照品,经过质量校正系数进行校正,用甲醇溶解,配制成质量浓度为1.00 mg/mL的内标储备液,于2~8 ℃保存;用乙腈-水(30∶70, v/v)稀释至25 ng/mL,作为内标工作液。
1.3 血浆样品前处理
向96孔板中加入50 μL血浆样品,然后加入10 μL 25.0 ng/mL内标工作溶液和10 μL 100 mmol/L醋酸铵水溶液,涡旋混匀;加入400 μL甲醇沉淀,振荡混匀,于4 ℃以4 000 r/min离心5 min;转移100 μL上清液于40 ℃吹干,用300 μL乙腈-水(30∶70, v/v)复溶,进样分析。
1.4 分析条件
色谱柱:Acquity UPLC BEH C8柱(50 mm×2.1 mm, 1.7 μm,美国Waters公司);柱温:40 ℃;自动进样器温度:5 ℃;流速:0.4 mL/min;流动相A:乙腈-水(95∶5, v/v);流动相B: 0.1%(v/v)甲酸水溶液(含5 mmol/L醋酸铵);进样量:5 μL。洗脱梯度见表1。
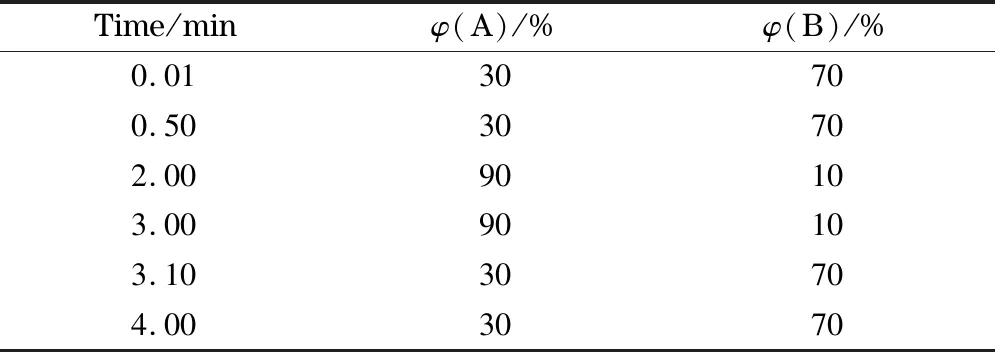
表1 梯度洗脱程序
A: acetonitrile-water (95∶5, v/v); B 0.1% (v/v) formic acid aqueous solution containing 5 mmol/L ammonium acetate.
离子源:电喷雾电离(ESI)源,正离子模式;扫描模式:多反应监测(MRM)模式;离子源温度(TEM): 550 ℃;离子化电压(IS): 2 500 V;碰撞气(CAD)压力:0.06 MPa;喷雾气(GS1)压力:0.4 MPa;辅助加热气(GS2)压力:0.4 MPa。苯海索反应检测离子对为302.3→98.0 (m/z),碰撞电压(CE)为32 V;苯海索-d11反应检测离子对为313.2→98.1 (m/z), CE为33 V;苯海索及苯海索-d11的去簇电压(DP)、入口电压(EP)和碰撞室出口电压(CXP)分别为100、10和13 V。苯海索及苯海索d-11的质谱图见图1。
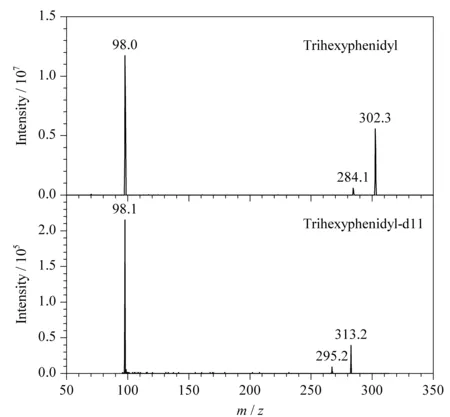
图1 苯海索和苯海索-d11的质谱图
1.5 临床试验方案
本次试验为随机、自身交叉、两序列两周期、设盲的人体生物等效性试验。以力生制药研制的盐酸苯海索片(2 mg)为受试制剂,以Watson生产的盐酸苯海索片(2 mg)为参比制剂进行空腹和餐后的单次口服给药的生物等效性相关试验。该临床试验在获得伦理委员会批准后开展进行。相邻两周期之间设置清洗期,时间为14 d。
研究分为两部分,第一部分为空腹给药,第二部分为餐后给药,两部分各筛选30例受试者。受试者充分了解实验内容后签署知情同意书。其中空腹试验中22号受试者因故主动退出,因此未纳入生物等效性分析。
试验每例受试者在每个周期服药前1 h内(记为0 h)及服药后15、30、45、60、80、100、120和150 min,以及3、4、6、8、12、24、36、48、72、96和120 h采血。所采集的血样置于含有肝素钠为抗凝剂的采血管中,采集完成后于1 h内于2~8 ℃以4 000 r/min离心10 min,获得血浆样品。
1.6 数据处理和统计分析
使用WinNonlin 8.0软件处理血药浓度数据并计算药代动力学参数,同时对参数进行统计分析。将达峰浓度(Cmax)、时间从零点到最后一个时间点的血药浓度-时间曲线下面积(AUC0-t)和时间从零点到无穷大时的血药浓度-时间曲线下面积(AUC0-∞)经对数转换后进行方差分析,并通过计算90%置信区间的方法对参比制剂和受试制剂进行生物等效性分析。
2 结果与讨论
2.1 方法学考察
2.1.1线性关系
取空白血浆950 μL,加入不同浓度的标准工作液50 μL,配制成苯海索质量浓度分别为0.100、0.200、1.00、2.00、5.00、15.0、35.0和40.0 ng/mL的血浆样品,按1.3节描述进行前处理并进样分析。在Analyst软件中积分,以待测物与内标峰面积的比值(y)为纵坐标、待测物的血药浓度(x, ng/mL)为横坐标进行线性回归,权重因子(w)为1/x2。结果表明,苯海索在0.100~40.0 ng/mL范围内,相关系数(r)为0.998 4~0.999 0,线性关系良好。
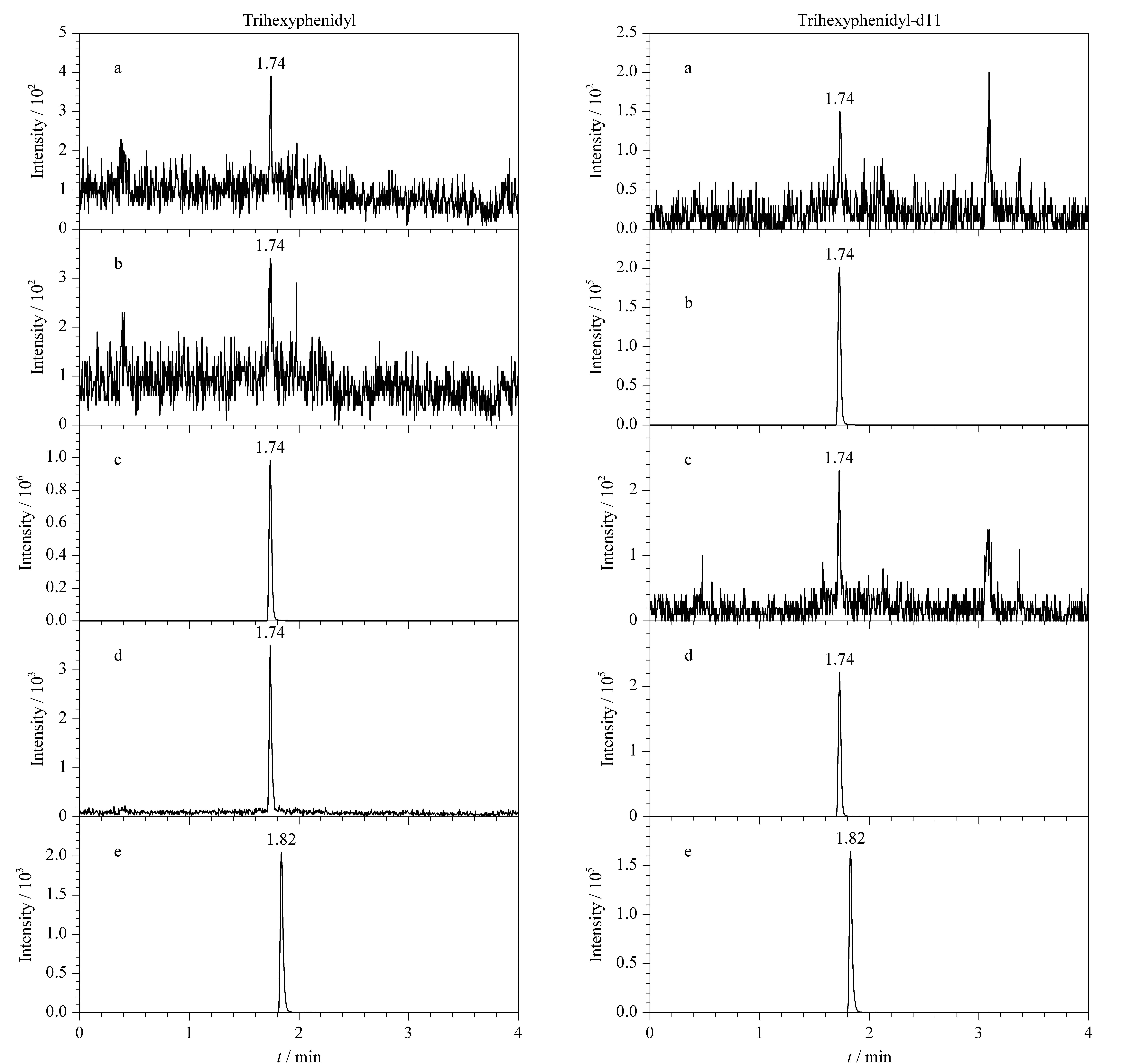
图2 人血浆中苯海索及苯海索-d11的典型提取离子色谱图
2.1.2准确度和精密度
取空白血浆950 μL,加入不同浓度的质控工作液50 μL,配制成定量下限(LLOQ)、低、中、高4个水平(0.100、0.250、2.00和30.0 ng/mL)的质控样品,每个水平各重复6个,按1.3节方法进行前处理,并连续测定3批,考察批内、批间的准确度和精密度。实验结果显示,批内和批间准确度分别为97.9%~115.5%和103.1%~110.5%,批内和批间精密度分别为2.5%~10.7%和4.5%~7.8%。表明方法准确度及精密度均良好。
2.1.3选择性
取6批不同来源的空白血浆,通过1.3节方法进行前处理(其中内标工作液以乙腈-水(30∶70, v/v)代替)后进样检测。取空白血浆、定量下限浓度血浆样品、定量上限(ULOQ)浓度(40.0 ng/mL)的血浆样品和健康受试者单剂量口服2 mg盐酸苯海索片1.0 h后的血浆样品,按1.3节方法进行前处理(其中含定量上限浓度苯海索的血浆样品中内标工作液用乙腈-水(30∶70, v/v)代替)后进样检测,相关色谱图见图2。实验结果显示,不同来源空白血浆中内源性物质对苯海索及苯海索-d11的测定无干扰,同时待测物和内标互相不干扰测定。
2.1.4基质效应
取来自6个受试者的空白基质,在低、中、高3个水平(0.250、2.00和30.0 ng/mL)下分别计算苯海索及内标的基质因子,并且计算经内标归一化后的基质因子。基质因子是指在基质中待测物的响应峰面积与纯溶液中待测物响应峰面积的比值,内标归一化的基质因子为待测物的基质因子与内标基质因子的比值。结果显示,高、中、低3个水平的质控样品经内标校正后的基质因子分别为99.0%、99.0%和101%,表明基质效应不影响苯海索的定量测定。
以高脂(含300 mg/dL甘油三酯)和溶血(将全血涡旋后加入对应体积的空白基质中混匀配制成2.0%(v/v)溶血血浆)基质配制低水平(0.250 ng/mL)和高水平(30.0 ng/mL)血浆样品,通过1.3节方法进行前处理后进样分析,用于考察高脂和溶血对于样品检测的影响。结果显示,高脂及溶血均不影响测定方法的准确度和精密度。
2.1.5提取回收率
将低、中、高3个水平(0.250、2.00和30.0 ng/mL)的待测物苯海索样品进行前处理,与未经提取处理的样品进行比较,考察回收率。结果表明,低、中、高3个水平血浆样品中苯海索的提取回收率分别为98.4%、95.2%和94.9%,显示该蛋白质沉淀的前处理方法提取回收率较高。

表2 健康受试者空腹(n=29)及餐后(n=30)服用受试制剂及参比制剂后的主要药代动力学参数 (mean±SD)
Tmax: time ofCmax;Cmax: maximum plasma concentration; AUC0-t, AUC0-∞: areas under the plasma concentration-time curves from zero to the last measurable concentration or to infinity;t1/2: terminal half-life.
2.1.6稳定性
实验分别考察了低含量(0.250 ng/mL)和高含量(30.0 ng/mL)血浆样品的冻融稳定性、长期储存稳定性、前处理过程中的稳定性和全血稳定性。取在不同实验条件下放置的样品,通过1.3节前处理方法处理后进样分析。实验结果显示,血浆样品于-80 ℃经过4次冻融、室温条件下放置26 h、处理后在进样室(2~8 ℃)放置7 d、于-80 ℃保存77 d,低浓度质控样品及高浓度血浆样品对比理论值的相对误差为-4.6%~10.5%,表明在上述条件下苯海索血浆样品均稳定。
配制含待测物的全血样品,部分全血样品离心获得血浆作为零时间点样品,其余全血样品在室温和冰浴条件下分别放置2 h后离心获得血浆作为全血稳定性样品,将稳定性样品与零时间点样品经过1.3节样品前处理后进样分析,结果显示全血样品冰浴及室温放置2 h内均稳定。
2.2 血浆样品中苯海索的测定
采用上述方法测定健康受试者在空腹和餐后单次给予盐酸苯海索参比制剂和受试制剂(2 mg)后的血药浓度,以时间为横坐标、血药浓度为纵坐标分别绘制参比制剂和受试制剂在空腹和餐后的苯海索血药浓度-时间曲线。受试者空腹及餐后单剂量口服盐酸苯海索片后前24 h平均血药浓度-时间曲线见图3。

图3 受试者空腹及餐后单剂量口服盐酸苯海索片后前24 h的平均血药浓度-时间曲线
如表2所示,采用WinNonlin 8.0软件,选择非房室模型,按实际采血时间及血药浓度计算主要药代动力学参数。分别将Cmax、AUC0-t和AUC0-∞对数转换,计算获得其90%置信区间。空腹试验中Cmax、AUC0-t和AUC0-∞的90%置信区间为82.2%~99.4%、82.3%~97.3%和83.4%~97.9%,餐后条件下分别为100.8%~122.8%、96.8%~112.4%和96.6%~112.1%,均在80.0%~125.0%范围内,表明参比制剂和受试制剂在空腹和餐后条件下吸收速度和吸收程度均无显著差异,具有生物等效性。
通过对比餐后和空腹试验结果,进食对于Cmax起到降低作用,并且达峰时间获得较大延长。而对于AUC0-t和AUC0-∞并无显著差异。因此,进食较大地影响了盐酸苯海索的吸收速度,而对于吸收程度并无较大影响,食物对于苯海索体内浓度的波动具有较大影响,参比制剂和受试制剂受食物影响的体内行为相似。
3 结论
本文建立了灵敏、高效、准确的UPLC-MS/MS法定量分析人血浆中的苯海索,并应用于空腹和餐后苯海索片人体生物等效性评价。该方法灵敏度高,前处理方法简便快捷,可应用于大批量样品分析,为仿制药的质量研究和控制提供参考。
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中老年保健(2021年3期)2021-08-22 06:52:12
恋爱婚姻家庭(2021年17期)2021-07-27 04:03:45
中国特种设备安全(2021年12期)2021-04-26 14:37:00
中国现代医药杂志(2020年10期)2020-12-14 07:20:06
天津医科大学学报(2019年3期)2019-08-13 06:53:16
中成药(2018年6期)2018-07-11 03:01:32
小学生导刊(低年级)(2016年8期)2016-09-24 07:56:23
中国粮油学报(2016年5期)2016-01-23 02:45:06
现代检验医学杂志(2014年1期)2014-02-06 01:29:48