
Sphk1拮抗剂的研究进展
2019-11-05 11:34:16杨皓然杨新美刘爱芹姚庆强
食品与药品 2019年5期
杨皓然 ,杨新美,刘爱芹 ,刘 波 *,姚庆强 *
(1.济南大学 山东省医学科学院 医学与生命科学学院,山东 济南 250200;2.山东省医学科学院药物研究所,国家卫生部生物技术药物重点实验室,山东省罕少见病重点实验室,山东 济南 250062;3.山东第一医科大学第一附属医院(山东省千佛山医院) 药学部,山东 济南 250014)
细胞内鞘脂代谢产物如神经酰胺(ceramide,Cer),鞘氨醇(sphingosine,Sp)和鞘氨醇-1-磷酸(sphingosine-1-phosphate,S1P)(见图1),是重要的信号分子[1],在调控细胞增殖、存活、迁移、分化及程序性凋亡等生命活动中发挥着重要作用。Cer和Sp是细胞增殖的负调控因子,能抑制细胞生长、促进细胞凋亡,而S1P则是重要的存活因子(survival factor),能促进细胞生长、抑制细胞凋亡[2]。细胞内Cer,Sp和S1P通过主要由鞘氨醇激酶1(sphingosine kinase 1,SphK1)参与的酶促反应维持动态平衡。已有研究表明,SphK1的异常与多种疾病的发生有关,包括镰状细胞病[3]、癌症[4]、动脉粥样硬化[5]、哮喘[6]等。近年,SphK1在癌症发生和发展中的重要作用被逐渐揭示,针对SphK1靶点的药物设计、开发也成为国内外研究的热点[7]。本文就SphK1的结构、功能及在研SphK1拮抗剂进行综述,期望为新型SphK1拮抗剂的研究开发提供参考。
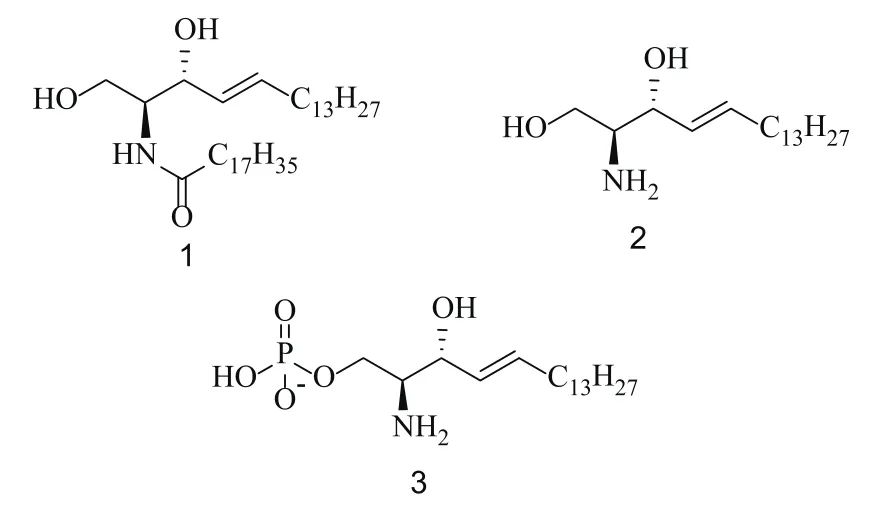
图1 Cer(1),Sp(2)和S1P(3)的结构
1 SphK1及其促进细胞增殖机制
SphK是一种高度保守的ATP依赖脂质激酶,由Olivera等于1998年首次从大鼠肾脏细胞中提取[8]。目前,在哺乳动物组织中发现有SphK1和SphK2两种亚型,其在酵母和植物中也均有表达[9-10]。人的SphK1基因位于17号染色体(17q25.2)上,相对分子质量(Mr)为42×103,主要分布于脑、心、脾、肺、肾及胸腺细胞中。SphK2基因位于19号染色体(19q13.2),SphK2比SphK1多200个氨基酸,Mr为63×103,主要分布于肾脏和肝脏肝细胞中[11]。SphK1和SphK2都有C1~C5共5个保守序列,具有高度同源性,在N端和C端分别有47 %和43 %的序列完全相同[12]。SphK1的主要功能是磷酸化Sp生成S1P,从而促进细胞增殖和血管生成,抑制凋亡。SphK2 的具体作用还不十分明确,通过RNAi技术干扰HEK293细胞的SphK2,能阻断血清撤离或药物诱导的细胞凋亡[13],但近期也有研究发现,选择性抑制SphK2的活性对肝癌[14]、多发性骨髓瘤[15]等多种癌症有一定的治疗作用,由于其作用机制的复杂性,针对 SphK2的拮抗剂还在研究中。
2013 年,人类SphK1晶体结构被解析出来,为基于靶点结构的合理药物设计提供了有力的科学依据[16]。人类SphK1(见图2)是含有384个氨基酸残基的蛋白质,包含9个α螺旋,17个β折叠和1个310螺旋。SphK1可分为两个结构域,其中,N-末端结构域(N-terminal domain,NTD)含6个α螺旋(α1~ α6)和6个β折叠(β1~β5,β17)。NTD的中心部位是4个平行β-折叠(β2,β1,β3,β4)和2个反平行β-折叠(β5,β17),β-折叠的两侧分别被α2,α3,α4和α1,α5,α6包围。C-末端结构域(C-terminal domain,CTD)是底物结合域,含有3个α螺旋(α7~α9),1个310-α螺旋和11个β折叠(β6-β16)。CTD中心部位的5个反平行β折叠(β7,β6,β16,β13,β9)位于CTD背面,另外6个反平行β折叠(β15,β14,β8,β10,β11,β12)位于CTD的前面,3个α螺旋(α7~α9),310-α螺旋和1个长的卷曲环区(coiled loop)分别位于11个β折叠的前面和背面,因此SphK1的配体结合域整体上呈一个“J形”口袋(见图2)。
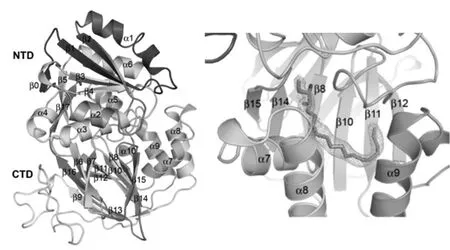
图2 SphK1的结构和“J形”疏水口袋
人体内鞘磷脂(sphingomyelin)的代谢途径为:细胞膜外的鞘磷脂(sphingomyelin)通过水解反应转化为Cer,然后Cer通过去酰化作用生成Sp,最后SphK1磷酸化Sp生成S1P。游离S1P可快速被S1P磷酸酶(S1P-phosphatase,SPP)降解成Sp或被S1P裂解酶(S1P lyase)不可逆地降解为软脂醛(palmitaldehyde)和磷酸胆碱(phosphoryl choline)。目前的研究认为S1P发挥生物学功能主要通过两种方式,一种为行使细胞外第一信使功能,即S1P以自分泌或旁分泌的方式分泌到细胞外,与G蛋白偶联受体上的相应S1P受体(S1P receptor,S1PR)结合产生“由内而外”的信号通路,进而调控细胞内多种生物学功能[17-18]。另一种为行使细胞内第二信使功能,即胞内S1P直接作用于细胞内的不同蛋白靶点介导多种生物学效应[19-20](见图3)。
2 SphK1拮抗剂

图3 Sphk1-S1P-S1PR信号通路
SphK1小分子拮抗剂的开发研究始于2003年[19],其作用机制多数是通过与SphK1的“J形”口袋结合来调节SphK1的功能[7]。目前在研的SphK1拮抗剂根据其结构可分为鞘氨醇类和非脂质类。近年,一些天然产物也被发现具有抑制SphK1的作用。本文总结了近年具有代表性的SphK1拮抗剂,并将各拮抗剂汇总见表1。

表1 SphK1拮抗剂汇总
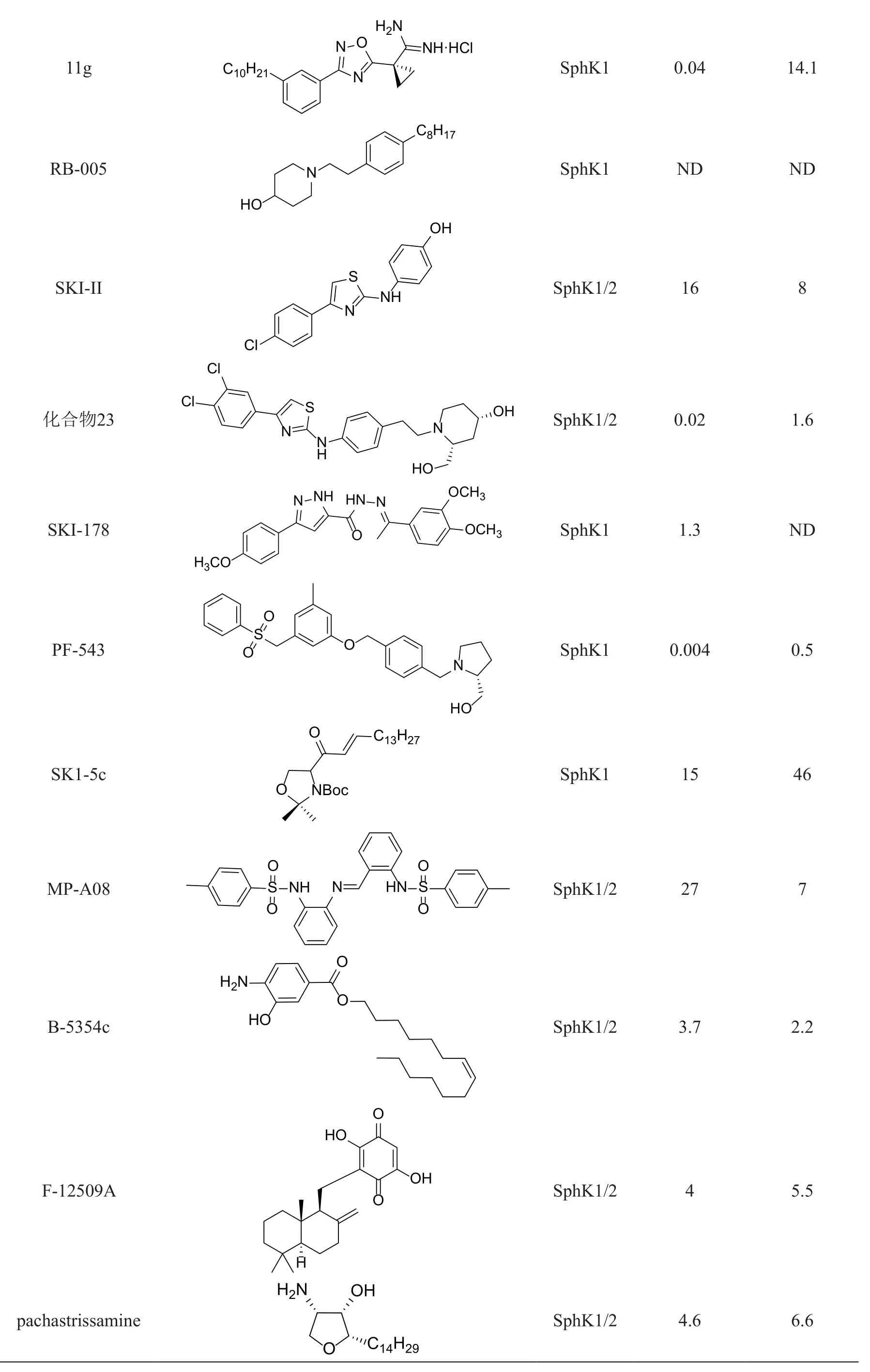
注:ND:未测定
2.1 鞘氨醇类SphK1拮抗剂
鞘氨醇类SphK1拮抗剂可看作是Sp的衍生物或类似物。该类拮抗剂主要针对Sp的极性头部、连接链、疏水尾部3部分进行结构修饰、改造(见图4)。
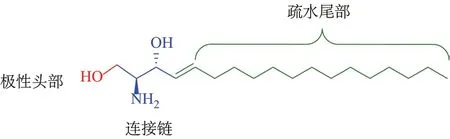
图4 鞘氨醇结构及其关键化学区域
SK1-I(BML-258)是同时对Sp 的疏水尾部和连接链进行改造而得到的化合物,也是第一个水溶性选择性SphK1拮抗剂。与通常在聚乙二醇或DMSO中递送的其他鞘氨醇类拮抗剂不同,SK1-I具有优异的溶解性,可以盐水溶液的形式给药[7]。SK1-I能选择性抑制SphK1的活性,对SphK2及其他激酶无抑制作用[21]。Sk1-I在白血病和U937细胞中可通过影响细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)和Akt信号来有效抑制肿瘤生长[21],在乳腺癌细胞中,Sk1-I可降低胞内S1P水平,也发挥出一定的抗增殖活性[22]。最近也有研究表明Sk1-I能通过提高抑癌蛋白TP53的活性而调控细胞周期和细胞凋亡[23]。
Sp的极性头部OH部分能被SphK1磷酸化后发挥信使作用,因此去除OH可能获得活性较好的SphK1拮抗剂,基于该思路得到了化合物Enigmol[24]。Enigmol不能被SphK1磷酸化,且对多种癌细胞表现出有效的抗癌活性,在小鼠结肠癌和前列腺癌的异种移植瘤模型中,口服给予Enigmol对两种癌细胞均显示出显著的生长抑制[25]。后续研究发现,对Enigmol进行结构优化后得到的化合物对多种癌细胞也具有良好的抗增殖活性[24]。于Enigmol的长链疏水尾部引入两个氟原子,得到CF2-Enigmol,其在小鼠体内实验中除了在抗增殖方面较亲代具有更好的效果,其在肺、脑和肾中具有较高的血药浓度,因此对于治疗肺、脑、肾组织中的实体瘤具有重要的意义[26]。
对Sp的连接链的氨基部分进行改造,分别得到化合物Dimethylsphingosine(DMS)和LCL351。DMS是SphK1、SphK2的共同拮抗剂,对蛋白激酶C(protein kinase C,PKC),神经酰胺激酶 (ceramide kinase,CK)和磷脂酰肌醇-3-激酶(phosphatidylinositol 3-kinase,PI3K)等多种重要蛋白激酶都具有抑制作用,选择性较低。DMS在多种癌细胞中均可诱导细胞凋亡[27],但也已被证实在较低浓时反而会增强SphK1的活性[28],另外,红细胞溶血和肝毒性等副作用也限制了其在临床上的应用[29]。LCL351是选择性的SphK1拮抗剂,在炎症性肠病的小鼠模型中,LCL351可选择性地抑制SphK1的活性而减少炎症物的产生,且在体内未见任何不良反应[30]。但作为抗肿瘤药,LCL351在体内生物利用度不高,药理活性也并不理想[31]。
Safingol可看作是Sp的疏水尾部双键还原后得到的化合物,也能同时抑制SphK2、PKC和CK等激酶的活性,选择性较低[29]。在结肠癌细胞中,Safingol通过引起细胞自噬抑制癌细胞增殖[32],对患有晚期实体瘤的患者进行的I期临床试验表明,Safingol可安全地与顺铂联合给药,仅显示出可逆的剂量依赖性肝毒性[33]。
近年,基于计算机辅助药物设计和构效关系研究设计合成的SphK1拮抗剂也在蓬勃发展。为了获得选择性更高的化合物,在Sp的基本骨架上引入脒基,由此得到了一系列脒类化合物,该类化合物对SphK1的选择性高于SphK2,并且对人PKC等多种激酶均无抑制作用[34]。VPC96091在纳摩尔浓度下即可显著降低U937细胞胞内S1P的水平,升高Sp和Cer水平,但仅在较高浓度(10 μmol/L) 时才会对该细胞中的ERK和Akt信号传导产生影响[7],抗增殖作用并不理想。在脒基的基础上,继续引入其他官能团如噁唑,得到化合物11g,该化合物对于SphK1的选择性是SphK2的705倍,是目前已知的选择性最好的拮抗剂[35]。虽然该类化合物的选择性和活性都很出色,但普遍存在体内代谢稳定性差,半衰期较短的缺点[34-35],且该类化合物的药动学性质还有待进一步研究[36]。基于构效关系研究设计合成的化合物RB-005在肺动脉平滑肌细胞中可降低SphK1基因的表达,同时可诱导人肺动脉平滑肌细胞中SphK1蛋白酶体降解,可作为SphK1拮抗剂研发的理想先导物[37]。
2.2 非脂质类SphK1拮抗剂
SKI-II是一类最早发现的非脂质类拮抗剂,对SphK1、SphK2都具有抑制活性,对PKC,ERK和PI3K等激酶无抑制作用,并能通过降低细胞内S1P的水平,升高Cer和Sp的水平,诱导多种癌细胞凋亡[38-39],此外,SKI-II还被证实可增强多柔比星等几种抗癌药物对癌细胞的作用效果[40-41],用SKI-II处理细胞也引起SphK1蛋白酶体降解,这有助于增强SKI-II降低细胞内S1P的能力[42]。在溃疡性结肠炎小鼠模型中,SKI-II有良好的口服生物利用度,但“脱靶”(off-target)效应限制了其在临床中的应用[43]。
美国的安进(Amgen)公司基于构效关系研究将SKI-II和Sp 的骨架进行拼接,开发了一系列高效的鞘氨醇激酶拮抗剂,代表化合物为化合物23,该化合物具有良好的物理化学和药动学性质,小鼠体内实验发现,虽然该化合物可有效抑制SphK1的活性,但并不能有效抑制肿瘤细胞的增殖,更深层的机制还有待进一步的研究[44]。
SKI-178是对SKI-II的同系化合物SKI-I进行结构修饰而产生的,对SphK1、SphK2都具有抑制活性。SKI-178在较低浓度时即可诱导多种癌细胞凋亡[45],目前还没有证据表明SKI-178会对PKC等蛋白激酶产生抑制作用。HL-60细胞中,SKI-178可加速促存活因子Bcl-2,Bcl-xl 和 Mcl-1的降解而发挥抗增殖作用[45]。AML细胞中,SKI-178除抑制SphK,还可破环细胞中的微管网络,从而诱导AML细胞的凋亡[46]。SKI-178在AML小鼠模型中具有良好的耐受性,安全有效,可作为多靶点抗癌治疗剂进一步发展[46]。
PF-543是高选择性的SphK1拮抗剂,对SphK1的选择性是SphK2的130倍,与RB-005和SKI-II一样,PF-543在发挥抑制作用的同时,也同时诱导SphK1蛋白酶体降解[47]。PF-543对原发性结直肠癌细胞具有抗增殖和细胞毒活性[48]。长时间较高剂量下PF-543可通过引起细胞自噬来诱导头颈部鳞状癌细胞的凋亡[49]。PF-543在镰状细胞性贫血小鼠模型中有效阻断镰状细胞、溶血和炎症的产生,并在小鼠肺动脉高压低氧模型中对心脏具有一定的保护作用[50]。除了发挥抗肿瘤作用外,PF-543在对抗炎症和自身免疫疾病方面的重要作用也应得到重视。
SK1-5c是一种选择性SphK1拮抗剂,对PKC等蛋白激酶均无抑制作用[51]。在结肠癌细胞中,SK1-5c可降低Akt信号的传导,抑制结肠癌细胞的增殖[52],SK1-5c也可诱导MDA-MB-231和MCF-7乳腺癌细胞凋亡,并在小鼠MDA-MB-231异种移植瘤模型中使肿瘤细胞的增殖受到抑制[53]。虽然SK1-5c是选择性SphK1拮抗剂,但其对其他脂质激酶、蛋白激酶和其他鞘脂代谢酶的影响需进一步研究。
MP-A08是通过计算机对接筛选得到的ATP竞争性SphK拮抗剂[54],其对SphK1和SphK2均有抑制作用。MP-A08不抑制人神经酰胺激酶(ceramide kinase,CK)和二酰基甘油激酶(diacylglycerol kinase,DAGK)等激酶的活性,并能降低S1P的水平,升高Sp和Cer的水平,但其并不诱导SphK1蛋白酶体降解。在小鼠肺癌异种移植瘤模型中,MP-A08能和ATP竞争鞘氨醇激酶上的结合位点,进而阻断促增生信号通路,减少肿瘤血管的形成和诱导细胞凋亡[54]。由于MP-A08相对于其他已知的SphK1拮抗剂有不同的作用模式,因而可为靶向SphK1的抗癌药物的研究提供新的思路。
2.3 天然产物类SphK1拮抗剂
天然产物一直是药物和药物先导物的重要源泉。目前发现一些从细菌和真菌中分离得到的天然产物,对鞘氨醇激酶也具有抑制作用。从海洋细菌中分离得到的B-5354c,可同时抑制SphK1和SphK2的活性,并能诱导前列腺癌细胞(PC-3)凋亡,在原位异种移植的PC-3细胞模型中与喜树碱发挥协同作用,增强喜树碱的抗肿瘤活性[55]。从真菌Trichopezizella barbata的培养液中分离得到的F-12509A,可同时抑制SphK1和SphK2的活性;HL-60细胞中,F-12509A可通过抑制SphK1来阻断S1P的产生,且无论多药耐药(multidrug resistance,MDR)基因表达如何,F-12509A均能在低浓度促进下诱导HL-60细胞凋亡[56]。虽然天然产物对SphK的选择性不太好,但它们可作为潜在的先导物用于开发更有效和特异性的SphK1拮抗剂。
天然产物 pachastrissamine(又名Jaspine B)是2002年及2003年从海绵中分离得到的一类无水鞘氨醇类化合物[57-58],其在结构上可看作 Sp的分子内脱水产物。佳司派恩(Jaspine B)具有顺式三取代的四氢呋喃环骨架,能同时抑制SphK1、SphK2的活性并对人类P388、A549、HT29、MEL28等多种癌细胞有细胞毒活性[59-60]。目前Jaspine B及其2,3,4-位异构体已全部合成出来[61],在测试中发现,其2位异构体对SphK1的选择性最高,可作为先导物进行改造、优化。目前,本课题组正利用分子对接等合理药物设计方法对2-位异构体进行衍生物设计、合成及生物活性研究,期望得到靶向SphK1的抗肿瘤临床候选药物。
3 结语与展望
SphK1与肿瘤等疾病之间联系密切,可作为癌症治疗的理想靶标。近年,SphK1拮抗剂发展迅速,特别是计算机辅助药物设计和构效关系研究的兴起,为这类药物的设计提供了更加科学的指导。但在研 SphK1 拮抗剂的选择性、体内活性或药动学性质限制了其临床应用,目前还没有上市及进入临床试验的候选药物,因此设计开发对 SphK1 具有高亲和力的特异性拮抗剂具有广阔的发展空间和重要的临床意义。
4 致谢
感谢山东省医学科学院院级科技计划(2016-5),山东省医学科学院医药卫生科技创新工程经费支持。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
奥秘(创新大赛)(2019年9期)2019-10-09 02:03:56
小哥白尼(趣味科学)(2019年1期)2019-04-12 00:23:56
奥秘(2017年5期)2017-07-05 11:09:30
海南医学(2016年8期)2016-06-08 05:43:00
环境与生活(2016年6期)2016-02-27 13:47:10
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国社区医师(2015年14期)2015-12-24 00:37:31
生殖医学杂志(2015年11期)2015-02-28 16:32:16
