
染色体微阵列芯片分析技术在精神运动发育迟缓患儿中的应用价值
2019-11-04 08:43:18谢巧玲叶燕霞李艳霞梁福杨均秀辛晶
中国中西医结合儿科学 2019年5期
谢巧玲, 叶燕霞, 李艳霞, 梁福, 杨均秀, 辛晶
精神运动发育迟缓是指包括运动、语言、认知、社会交流、日常活动能力2个以上发育能区显著落后于同龄儿。如不进行早期干预,多数发展成智力低下,是儿童常见神经疾患之一[1]。导致发育迟缓的病因复杂,遗传异常是其重要因素,占30%~60%[2]。染色体微阵列芯片分析技术(array-based comparative genomic hybridization,aCGH)是一种高分辨率的全基因组检测技术,可检测拷贝数变异(copy number variants,CNVs)的微小缺失和重复[3]。本研究旨在探讨aCGH技术的临床价值。
1 资料与方法
1.1 临床资料 选择2015年1月至2019年6月在我科就诊的精神运动发育迟缓患儿150例,其中男87例,女63例;年龄2个月至14岁,平均(2.00±0.35)岁。
1.2 诊断标准 参照《常见中枢性运动发育落后/障碍的规范化诊断》中精神运动发育迟缓的诊断标准[4]。
1.3 纳入标准 (1)符合精神运动发育迟缓的诊断标准;(2)年龄2个月至14岁;(3)患儿家属知情同意,且获得医院伦理委员会批准。
1.4 排除标准 (1)其他任何可导致脑功能紊乱的严重躯体或精神疾患;(2)明确头颅外伤史;(3)服用导致认知功能损害药物者。
1.5 方法 严格遵循Affymetrix公司提供的操作流程。采患儿静脉血2 mL。按试剂盒说明书操作,先后加入Protease K溶液20 μL、样本200 μL及缓冲液AL 200 μL,震荡混匀后56 ℃温育10 min,加入200 μL无水乙醇,漂洗2次;加入配制好的消化试剂消化,随后加入配制的连接试剂。连接采用QIAamp DNA Blood Mini Kit(Qiagen,德国)试剂盒提取标本基因组DNA,利用紫外分光光度计检测基因组DNA的浓度和纯度对标本进行质量控制,保证DNA浓度>50 mg/L,A260/A280在1.8~2.0。进行PCR扩增、提纯、定量,加入片段化试剂进行DNA片段化处理;将DNA溶液标记后加入杂交试剂95 ℃ 10 min后49 ℃孵育至少1 min,加200 μL处理好的样本载入到芯片中,并置密闭杂交盒中50 ℃ 16~18 h进行杂交反应;洗染芯片后扫描分析。取5 μL基因组DNA用于染色体微阵列分析(chromosomal mlcroarray analysis,CMA)检测(美国Affymetrix CytoScan 750K芯片平台)。该芯片含有350万个探针(195万个拷贝数探针和75万个SNP探针),除了加密覆盖拷贝数多变区、遗传疾病关联区、基因分布区等以外,还均匀分布于人类基因组整个范围。以Affymetrix提供的正常人DNA作为对照标准。采用Affymetrix GeneChip System 3000Dx v.2芯片系统进行数据采集,Affymetrix 7G扫描仪扫描芯片,扫描信号图经Chas软件分析、计算。
2 结果
检测发现存在CNVs异常占16.7%(25/150),其中10例为已知综合征,Brunner综合征1例,Saethre-Chotzen综合征1例,Prader-Willi综合征1例,Williams-Beuren综合征2例,Klinefelter综合征2例,Siemencs综合征1例,Cohen综合征1例,22q11.2重复综合征1例,为6%(10/150)的患儿基本明确了病因诊断。16p13.11微缺失综合征2例,7q末端缺少(大片段缺失)1例。临床致病性拷贝数改变7例。临床意义不明5例。见表1。
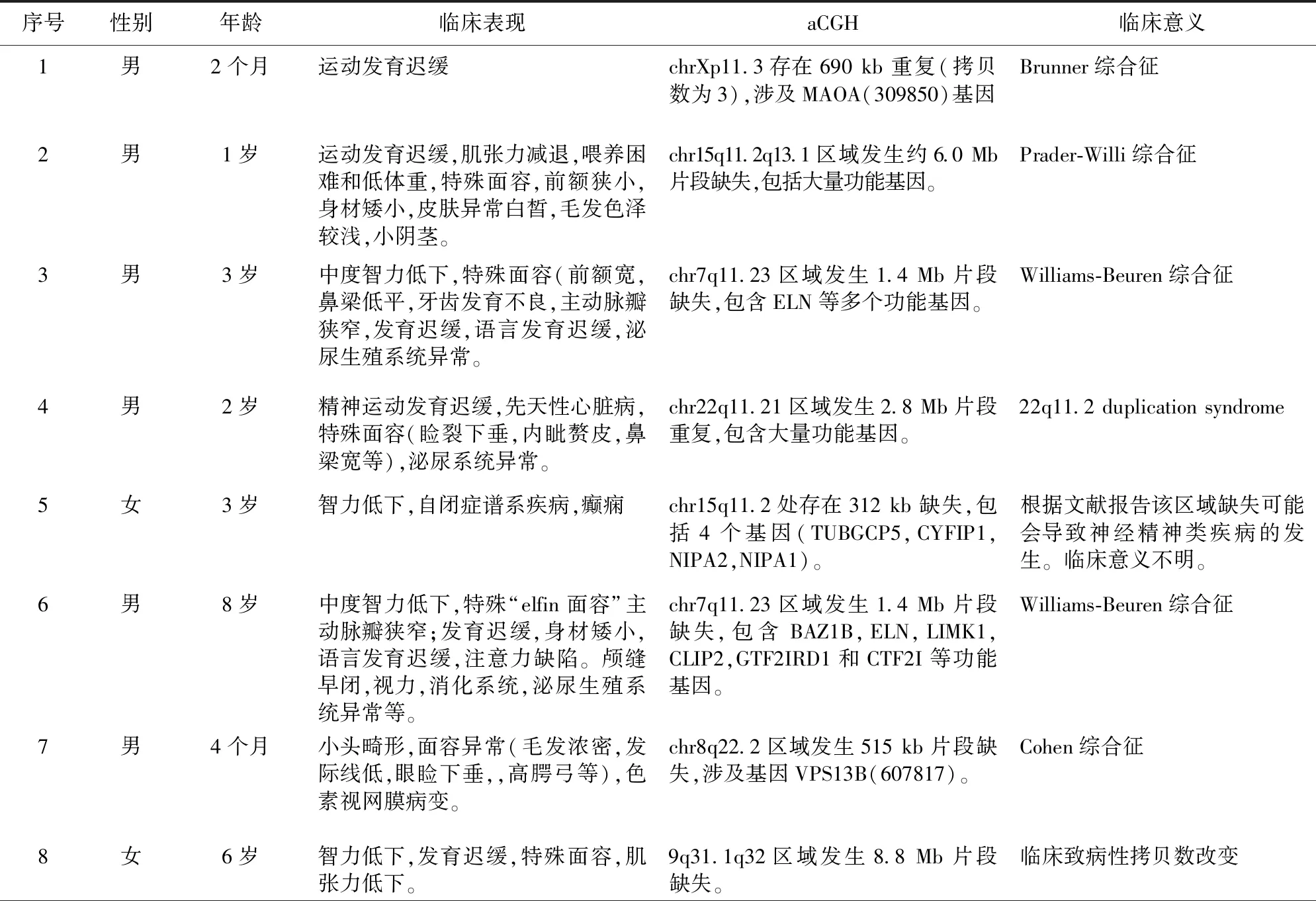
表1 25例患儿临床资料及aCGH结果
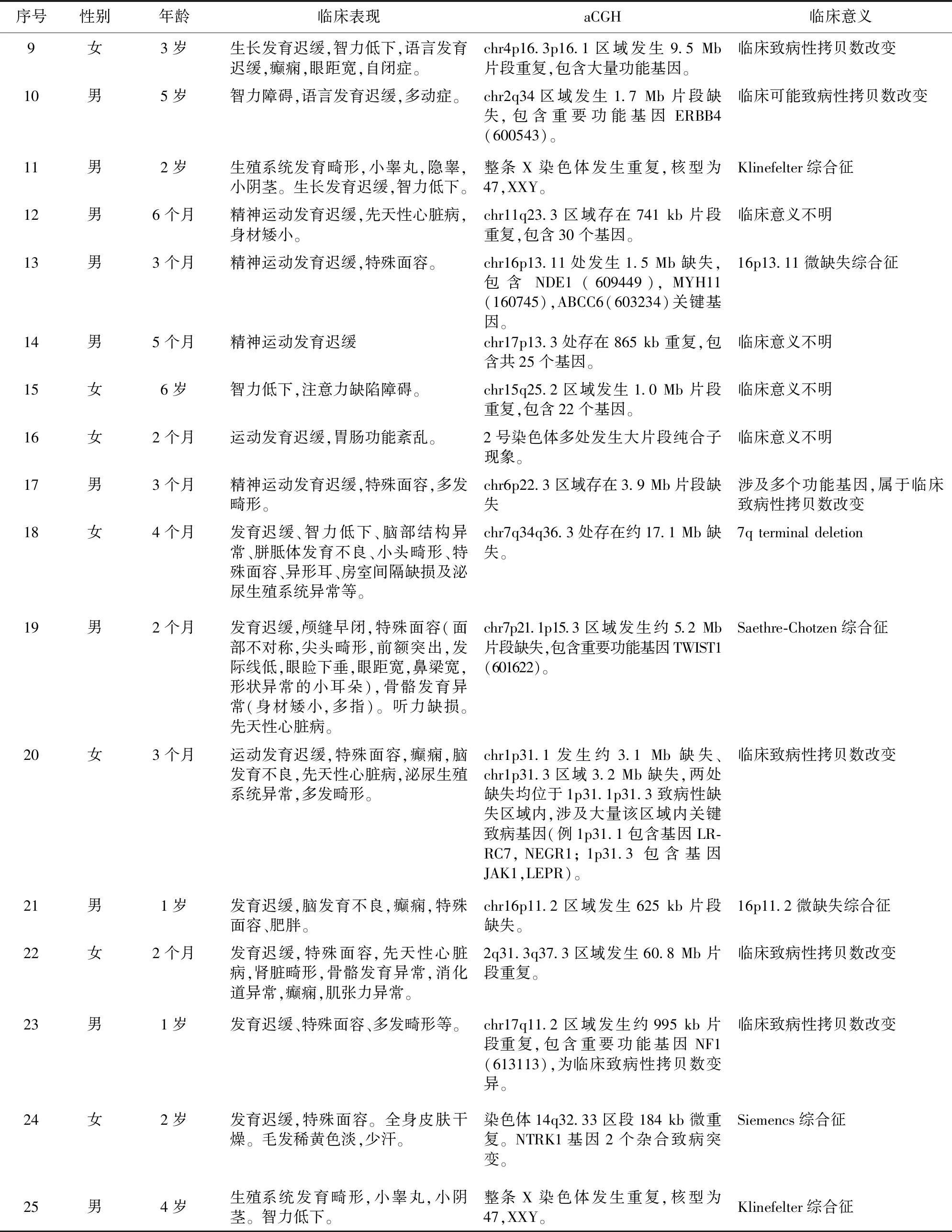
续表
3 讨论
精神运动发育迟缓是小儿常见的一种发育障碍,临床表现为运动及认知功能损伤,发病率为1%~3%[5]。本研究应用aCHG检测精神运动发育迟缓患儿150例,25例发现致病性CNVs改变,阳性率为16.7%。与张端秀等[6]报道的阳性率结论基本一致。因此提出CMA对于原因不明的发育迟缓/智力落后、非已知遗传性综合征的多发畸形等病因诊断是首选试验。阳性病例中,已知综合征有10例,而多数微缺失、微重复综合征没有被临床所认识。此外,本研究中有多个缺失或重复的片段大小≥3 Mb,可以认为是明确致病的。另外发现了7个数据库中暂未定义综合征或疾病区域的CNVs,考虑为临床致病性拷贝数改变。
Siemencs综合征是一组外胚层发育缺损的先天性疾患,累及皮肤及其附属结构如牙和眼,甚至累及中枢神经系统。通常以常染色体隐性遗传,涉及关键基因NTRK1基因突变[7]。携带致病基因突变的父母每次生育子女均有25%的可能为患者,患者父母的其他亲属亦有携带相同致病突变的风险。
Williams综合征是一种因7号染色体长臂11.23区域片段微缺失引起的先天性、神经发育障碍性疾病,发病率为1/7 500~1/20 000[8]。表现为“小精灵”面容、高钙血症,生长发育迟缓,智力低下,喂养困难、多系统异常、心理行为问题等。病情发展程度及预后因人而异,因弹力蛋白基因缺失引起心血管异常是导致患儿最终猝死的危险因素。
Klinefelter综合征是男性不育症常见的遗传学病因之一,占到无精症患者的11%,不育症患者的3%[9]。典型核型为47,XXY,还有46,XY/47,XXY及46,XX/47,XXY(嵌合型)等。临床主要表现为身材高大、生殖系统发育畸形、生殖能力低下,青春期发育延迟,第二性征发育异常,无精症,肌张力异常,器官过距、关节异常,可伴有语言障碍,学习障碍,精神分裂症等。早期识别需注意生殖器发育异常,成年男性患者不育、女性化乳房、性功能障碍等。Cohen综合征呈常染色体隐性遗传,其典型临床症状包括智力低下,产后小头畸形,面容异常,色素视网膜病变,近视,间断性中性粒细胞减少等[10]。
Brunner综合征为X连锁隐性遗传,受累男孩表现为中度智力低下,运动发育迟缓,自闭症,攻击性行为,学习困难,睡眠障碍等。
Saethre-Chotzen综合征,主要临床症状包括颅缝早闭,特殊面容(面部不对称,小头畸形,前额突出,发际线低,眼睑下垂,眼距宽,鼻梁宽,形状异常的小耳朵等),骨骼发育异常(如手脚骨骼畸形,身材矮小,并指)[11]。部分患者伴有发育迟缓,学习困难,听力缺损,先天性心脏病等症状。
Prader-Willi综合征,临床表现为发育迟缓,肌张力减退,喂养困难和低体质量[12]。儿童期食欲旺盛导致肥胖。患儿有轻度到中度的智力低下和学习困难,行为异常,睡眠障碍和特殊面容(前额狭小,嘴呈三角形)等。身材矮小,皮肤异常白皙,毛发色泽较浅,生殖系统发育异常。发病率为1/10 000~1/30 000。70%Prader-Willi综合征患儿是由于15号染色体该致病区域父源片段发生缺失,25%患儿是由于15号染色体该区域母源片段单亲二倍体所致,其余可能是由于印记基因的突变,属于生殖细胞形成过程中和胚胎早期发育的随机事件。
7q terminal deletion临床症状有发育迟缓、智力低下、脑部结构异常、胼胝体发育不良、小头畸形、特殊面容、唇腭裂、异形耳、先天性心脏缺陷(房室间隔缺损)及泌尿生殖系统异常等。chr16p11.2、16p13.11处微缺失综合征相关,主要临床表现包括:生长发育迟缓,智力低下,学习困难,癫痫,特殊面容等表现。与研究结论相符合,遗传自非典型患病亲代的CNV,位于染色体16p11.2、16p13.11也属于病理性的。
CMA为我们进一步了解发育迟缓患儿基因缺陷形成过程中的作用及相关机制奠定基础,也为我们寻找患儿基因诊断及进一步治疗方法提供新的方向。本研究中致病CNVs检出率为16.7%,包括已知综合征有10例,说明遗传因素是引起儿童智力低下的重要因素,基因异常特别是拷贝数变异是引起发育迟缓/智力低下的重要原因。但由于CMA存在不能检测平衡异位、倒位等缺点,因此临床医生根据受检者具体情况选择合适的检测手段,以达到利益最大化目的。患者首先应该进行CMA检查,CMA检测异常的患者还要进行FISH、G-显带、qPCR或多重探针连接扩增检测,以证实CMA的结果[13]。针对所发现的罕见CNV,尽可能采用双亲样本重复aCGH检测,了解其是否为新生CNV。
精神运动发育迟缓儿童,如不进行早期干预,3岁以后多数发展成智力发育障碍,严重者可致终身残疾。本研究提示临床应重视从婴儿期开始发育状况的监测和筛查。通过CMA,不论有无躯体畸形的发育迟缓的儿童,染色体正常的患儿有可能得到更多的遗传信息[14-16]。这将有利于早期干预、残疾预防、降低同胞的再发风险、家庭风险咨询、康复决策实施,也为其他疑难病例开辟了一条有效的遗传学诊断途径。
猜你喜欢
河北医学(2021年10期)2021-10-27 00:37:14
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
小天使·二年级语数英综合(2017年4期)2017-04-18 17:29:21
小天使·四年级语数英综合(2017年4期)2017-04-18 09:15:43
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
发明与创新(2015年25期)2015-02-27 10:39:16
少年文艺·开心阅读作文(2014年5期)2014-10-08 16:11:31
