
氨气还原氮化五氧化二钒制备V(N,O)粉体与机理研究
2019-10-28 02:09:58赵世强
有色金属科学与工程 2019年5期
赵世强
(北京科技大学钢铁冶金新技术国家重点实验室,北京100083)
近年来,由于高熔点、高硬度、高强度、良好的导电性和导热性等特性,过渡金属氮化物受到越来越多的关注[1-4].考虑到这些典型特性,它们已广泛应用于冶金、电子、催化剂等领域[4-6].其中,氮化钒是一种超导体,其转变温度范围为2~9 K[7].此外,研究者还发现氮化钒在催化加氢处理中表现出优异的活性和选择性,例如脱氢脱氮(HDN),脱氢脱硫(HDS)和脱氢脱氧(HDO)[8].
氮化钒一般是通过各种高温反应制备 (高于1623 K),例如V2O5或V2O3的碳热还原氮化,低温下镁热还原氮化[9,10].近年来,人们研究了一些制备氮化钒的新方法:气相分解,金属氧化物的氨解和自蔓延高温合成(SHS)[11-13].Bang等[11]通过微波等离子体分解气相原料(VCl4)成功合成了VN纳米粒子.Choi等[12]通过V2O5与纯NH3的程序升温反应获得VN粉末.尽管已经开发了一系列方法来生产氮化钒,但由于高成本和低产率,这些方法的工业应用受到限制.
由于具有相同的晶体结构,VO1-x和VN1-x难以通过XRD图谱区分,且VO1-x和VN1-x可以形成连续的固溶体.因此,除去氮化钒晶格中存在的所有氧原子是非常困难的.文献[14]已经通过NH3还原V2O5来制备VN,但他们没有显示最终产物的详细化学分析,也没有详细地研究其中的反应机理.因此研究V2O5与NH3在723~873 K的还原氮化过程中制备VN粉末,并详细研究整个过程中的反应机理.利用FE-SEM,TGA,XPS和BET进行材料表征,并测量最终样品的氮含量.V2O5的还原氮化过程可以用方程式(1)来描述.在还原过程中,详细的反应机理也将在下面的研究中讨论.
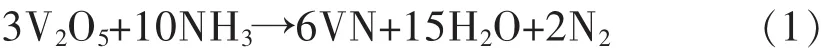
1 原料与实验过程
1.1 原料
V2O5和NH3用于制备VN粉体.其中,V2O5为分析级试剂,由Alfa Aesar提供,纯度高于99.6%.图1显示了V2O5粉末的X射线衍射图谱.
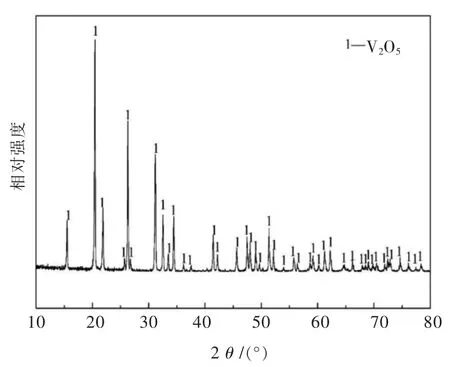
图1 五氧化二钒的物相谱Fig.1 XRD patterns of the experimental raw material V2O5
1.2 实验过程
实验装置由质量流量计 (Alicat 21-1-10-0-500-KM0410,美国)和热重分析仪(TG,HTC-2,北京恒久仪器有限公司,中国)组成,用于监测V2O5的重量变化,在还原过程中记录样品的TG曲线.在每次实验中,将100 mgV2O5粉末放在氧化铝坩埚(直径7 mm,高7 mm)中并放置在炉子的恒温区中.将炉温升至目标温度(773~873 K),加热速率为10 K/min.然后,将氨通入炉中,开始还原V2O5并持续一定反应时间.根据样品的TG曲线,当反应结束时,氨转换为高纯度氩(氧气含量低于0.0005%).然后,在氩气下将样品冷却至室温.在还原过程中,气体流速始终保持在75 mL/min,由气体质量流量控制器控制.
用 XRD(Model TTRIII,Japan)技术分析样品的物相组成.通过使用场发射扫描电子显微镜 (FESEM,ZEISS SUPRA 55)观察样品的形貌.通过比表面积方法 (BET,AUTOSORB-IQ2;Quanta chrome,Boynton Beach,Florida,USA)测量样品的表面积.采用 X 射线光电子能谱(XPS,AXIS U1TRA,Kratos,日本)分析表面的化学性质,用惰性气体熔融后的热导法测定最终样品的氮含量(GB/T 24583.2-2009).
2 结果与讨论
2.1 五氧化二钒的等温还原
在773 K,823 K和873 K下,V2O5粉末的还原进度随时间的变化曲线如图2所示.V2O5的还原进度可通过公式(2)计算得出.

其中,Wa和Wt分别是原始样品的实际重量损失和理论最大重量损失(从V2O5到VN).
从图2中可以发现,还原进度低于0.5的还原反应速率极快.当还原进度高于0.5时,发现还原速率受温度变化的影响.根据773 K,823 K和873 K下获得的最终样品的重量损失数据,重量损失率约为26%~28%,这个数值接近从V2O5转变成VN的理论重量损失值.众所周知,五氧化二钒的熔点较低(约为943 K).当温度达到其熔点时,V2O5会熔化形成液相并存在一定的挥发现象.因此,为了降低V2O5在高温下的挥发损失,没有研究更高的温度;此外,由于相同温度下气液反应速率远低于气固反应速率,低于V2O5熔点的温度更值得研究.相反在较低的温度下(723 K),还原反应速率太慢,不能达到还原的目的.
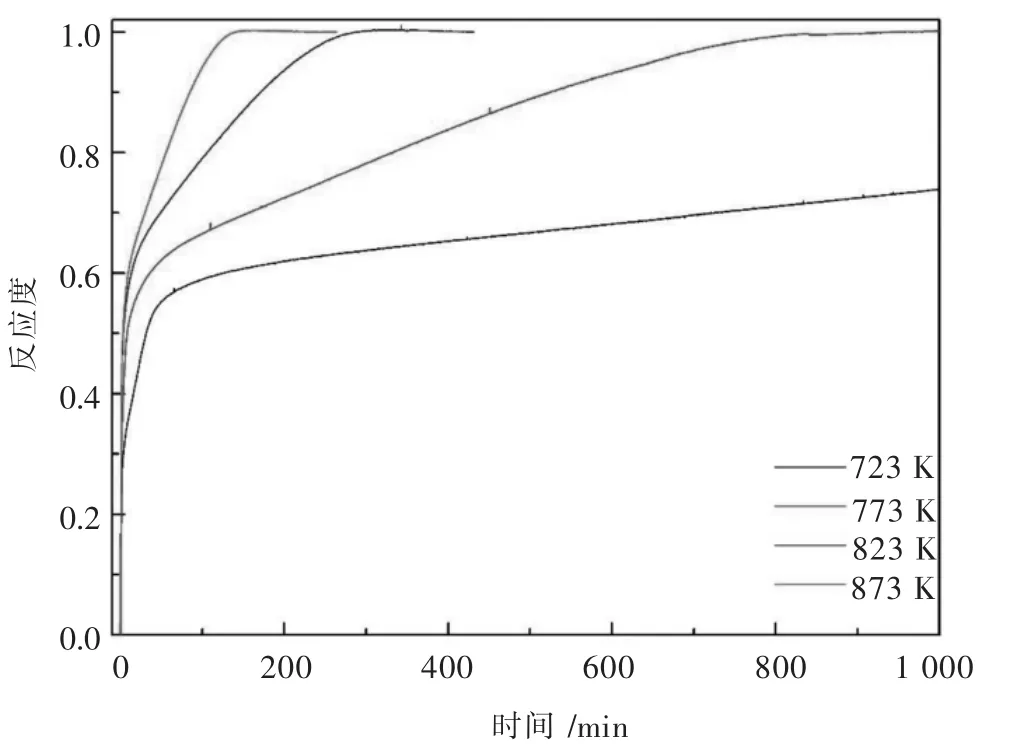
图2 不同温度下的还原动力学曲线Fig.2 The kinetic curves at different temperatures
2.2 物相分析
图3 不同温度下反应不同时间的样品物相谱Fig.3 The phase evolution of the samples prepared at different time
从图2中可以看到在723 K下反应是一直进行的,只是反应速率较慢,这是一个明显的动力学问题.鉴于773 K,823 K和873 K下物相演变以及图2中动力学曲线的一致性,在723 K下的物相转变过程与前者是一致的,只是由于动力学因素在反应后期反应速率较慢.因此,分析773 K,823 K和873 K下,反应不同时间的样品的物相图谱,如图3(a)~图3(c)所示. 图 3(d)是 VO2(JCPDS 82-661),V4O7(JCPDS 71-423),V2O3(JCPDS 84-317)和 VN(JCPDS 35-768)的标准图谱.通过与标准图谱对比,从图3(a)~图 3(c)中可以得出:VO2,V4O7和 V2O3作为中间产物在反应过程中形成.当反应时间为4 min时,图谱的所有峰都指向VO2;随着反应过程的进行,VO2峰强度降低,而V4O7的峰值明显增加;当时间为420 min时,VO2和V4O7彻底消失,所得样品由V2O3组成;最后,V2O3的峰值逐渐消失,在氨气氛下出现VN峰(JCPDS 35-768),而且未检测到其他杂质相.随着的温度的升高,物相转变速率越快,得到单一VN相时间越短.根据物相分析结果,V2O5与氨气在773 K至873 K范围内的物相转变顺序为:V2O5→VO2→V4O7→V2O3→VN.这表明当V2O5在 773 K至 873 K的温度范围内转变为VN时,晶体结构从正交立方体转变为面心立方体.从V2O5到VN的转变可以描述为反应式(3)~式(6).
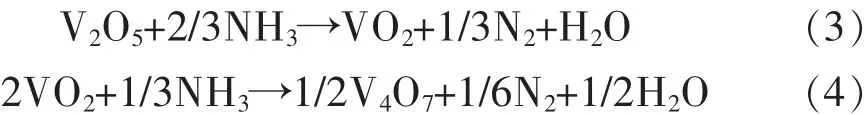
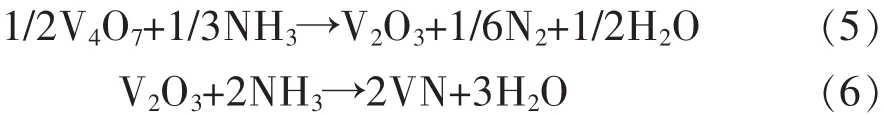
图4(a)所示为773 K,823 K和873 K下获得的最终产物的XRD图谱.在图4(a)中,可以明显地看到,几乎所有峰都为VN.为了确定较优反应温度,分析了在不同温度下合成的VN粉末的氮含量,结果如表 1 所列.图 4(b)为图 4(a)的局部放大图,如图4(b)所示,VN在823 K和873 K的特征峰略微偏移到略低的衍射角.结果表明,随着温度的升高,VN相的氮含量增加,这可能是由于氧原子的逸出,越来越多的氮原子占据了VN面心立方结构中氧原子的晶格位置.结合上述分析,在反应后期V(N,O)粉末中氧原子逐渐以水蒸气形式逸出并被氮原子取代.由于氮和氧原子的质量分数十分接近,分别为7和8,样品质量变化很小,很难利用热重分析仪得到的动力学曲线(图2)分析后期反应,其反应度接近于1.为了分析氨气还原氮化反应的后期过程,利用化学分析法对产品粉末氮含量进行测量.随着反应温度的升高,样品氮含量从16.4%增加到17.4%,这与XRD图谱的结果非常吻合.然而,根据理论计算,VN的理论氮含量为21.4%.因此,在不同温度下获得的最终产品应为V(N,O)粉末.
图4 不同温度下得到的最终样品的物相谱(扫描速率为1.5o/min)及局部放大图Fig.4 The XRD patterns of final productions prepared at different temperatures with a scanning rate of 1.5o/min

表1 不同温度下最终样品的氮含量Table 1 Nitrogen contents of the samples prepared with various temperatures
2.3 微观形貌分析
图5显示823 K下还原不同程度的样品的形态演变.如图5(a)所示,原料V2O5颗粒是无孔的,呈片状或棒状.与图5(a)相比,还原产物的大小和形状几乎保持不变;然而,随着还原的进行,表面粗糙度增加.图5(d)中VO2转化为V2O3过程的表面微观结构变化最大;当还原程度达到0.65左右时,产品表面松散多孔,微观结构的变化持续到VN形成.如图5(f)所示,在表面形成大量的小颗粒,这可能是因为在还原过程中氧原子持续以水蒸气的形式逸出.
图6显示773 K,823 K和873 K下的最终还原产物的FE-SEM照片.如图5所示,最终还原产物在不同温度下的尺寸和形状变化很小,与原料V2O5粉末相同;然而,从图6(b)中清楚地看到表面上存在许多颗粒.与图6(b)所示的晶粒相比,由于晶粒生长,在较高温度下表面上的晶粒变大(如图6(b)右和图 6(c)右所示),特别是在873 K温度下.由于系统具有降低总自由能的趋势,原子/分子的热运动导致许多小孔的合并形成大孔,也可以说成是颗粒的粒度增加.温度越高,原子/分子的扩散能力越快,晶粒尺寸越大.此外,在773 K,823 K和873 K下制备的最终样品的比表面积测量结果分别为29.405 m2/g,21.106 m2/g和15.482 m2/g.随着反应温度的升高,表面积相应减小,该结果与上述形态分析具有良好的一致性.
2.4 材料表征
图7显示773 K下制备VN样品在O 1s和V 2p区域获得的高分辨率XPS光谱,可以看出527.00 eV和536.00 eV之间的O 1s的峰值尺寸相当大,并且O1的光谱不对称,这证明表面氧可以认为是晶格氧,晶格氧羟基(OH-)和吸附氧(O2-)分别位于 529.5 eV、532.20 eV 和 533.00 eV[15-17].V(N,O)的 O 1s能量区包括2个峰,如图7(a)所示.位于532.14 eV的峰应该是羟基 (OH-),这可能是由于样品暴露在空气中.在530.4 eV处的峰值与晶格氧相对应,并可以认为是V(N,O).通过化学分析证明存在VN晶格中的氧原子,这可能是因为氧原子在低温下几乎不能被脱除.图7(b)显示了在773 K下制备的V(N,O)样品的V 2p区域的XPS光谱.位于517.3 eV的峰属于氧化钒中存在的V5+[16-22].然而,这种钒氧化物不出现在X射线衍射图中(图4),表明它们可能是由于高表面活性引起的轻微氧化.轻微氧化是不可避免的,这个现象也出现在文献[20-21]中.在514.2 eV处的峰值比VN峰值(513.5 eV)略高,但低于其他钒氧化物,包括 VO2(515.65 eV),V6O13(516.2 eV),V4O9(516.8 eV)[22].因此,可以认为位于514.2 eV的V 2p3/2峰应为 V(N,O).

图5 823 K下反应不同程度的样品的形貌演变Fig.5 Morphology evolutions of samples at 823 K for different reaction extents

图6 在不同温度下反应的最终样品的微观形貌Fig.6 FE-SEM photographs of the final reduction product under different magnifications and different temperatures
2.5 反应机理
通过分析整个过程的物相和微观结构演变,详细地研究氨气还原五氧化二钒过程中的反应机理.为简化分析,假设所有相的颗粒形状为球形.当温度高于773 K时,在V2O5还原为V(N,O)期间的相变和形态演变可以通过图8中所示的示意图描述,并解释如下:
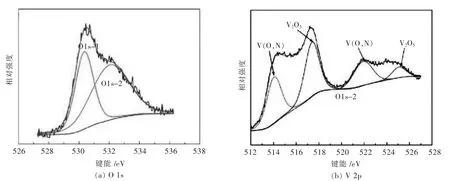
图7 873 K下反应得到的最终样品的XPS图谱Fig.7 The spectrum of XPS of the sample obtained at 873 K
1)当温度高于773 K时,将NH3气体引入炉中,原料迅速被还原为低价氧化物.结合XRD图谱的结果,V2O5可以快速的被还原成VO2和V4O7.在还原过程中,形成大量H2O并从反应界面逸出.在其他文献中,Kwon[14]分别在 503 K,593 K,663 K,763 K,923 K和1073 K研究了V2O5的还原过程.在他的研究中,也在593 K下发现了V4O9相的形成.然而,在本研究中,还原温度较高(773~873 K),这导致较高的反应速率,这使得难以观察V2O5和VO2之间的中间物相.然而,在VO2和V2O3之间发现了新物相V4O7.此外,他们发现在923 K时存在中间相VO0.9.本文中,在773 K下制备的最终产品的氮含量高达16.4%.

图8 氨气还原五氧化二钒制备V(N,O)粉体的反应机理Fig.8 Reaction mechanism of reduction of vanadium pentoxide when the temperature is higher than 773 K
2)随着反应程度的增加,VO2和V4O7相进一步被还原至V2O3.一部分V2O3也与氨气反应生成V(N,O).在还原过程中,从NH3分解的高活性H原子进入低价氧化物晶格并与O原子反应,导致氧含量降低.最后,可以得到产物V(N,O).在此过程中,很明显发现在表面上形成了大量的小颗粒.
3)众所周知,VO1-x和VN1-x具有相同的晶体结构并可形成固溶体,而且从VN晶格脱除所有氧原子难度大.在VN的晶格中,仍然存在少量氧原子并占据氮原子的位置.随着温度的升高,反应动力学进一步增加.氮原子更容易进入VN的晶格并占据氧原子的位置.这就是当温度升高时VN的氮含量增加的原因.
3 结 论
研究了773 K至873 K温度范围内氨气还原五氧化二钒制备V(N,O)粉体.利用XPS和XRD等手段,证实了773 K,823 K和873 K下制备的样品为V(N,O);此外,研究了物相和形态演变.具体结论为:
1)V(N,O)可以在773 K到873 K的温度范围内通过氨还原V2O5在短时间内成功制备.在固态还原过程中,VO2,V4O7,V2O3作为中间产物确实形成并且按照V2O5→VO2→V4O7→V2O3→V(N,O)的顺序进行还原.随着温度的升高,氮原子更容易进入VN晶格并占据氧原子的位置.最终产品的氮含量随温度的升高而增加.
2)在773 K至873 K的温度范围内,产品的颗粒尺寸和形状几乎与V2O5相同.但是,随着温度的升高,VN的晶粒尺寸增大.
猜你喜欢
发明与创新·中学生(2023年2期)2023-01-09 03:50:05
椰城(2021年12期)2021-12-10 06:08:52
陶瓷学报(2021年1期)2021-04-13 01:33:08
世界有色金属(2020年4期)2020-05-16 05:55:44
热处理技术与装备(2019年1期)2019-03-14 08:07:20
电子制作(2018年12期)2018-08-01 00:47:48
物理学报(2017年21期)2017-11-10 08:25:38
上海金属(2016年2期)2016-11-23 05:34:32
腐蚀与防护(2016年7期)2016-09-14 09:30:56
上海金属(2015年6期)2015-11-29 01:08:49
