
基于茎环法的RT-qPCR检测哺乳动物抗病毒siRNA的表达
2019-10-25 02:33:38李卫振任严新
复旦学报(自然科学版) 2019年5期
李卫振,任严新,李 杨
(复旦大学 生命科学学院,上海 200438)
病毒入侵会迅速激活宿主的抗病毒免疫反应,被感染的细胞能迅速发现病毒并作出反应.RNAi是植物系统性抵御病毒入侵的重要途径[1].RNAi在进化过程中高度保守,由内源或外源双链RNA诱导产生,能够高效特异性降解同源mRNA[2],产生大量长21~23bp的双链小RNA,切割后的siRNA 3′末端往往含有2个碱基的粘性末端[3].RNAi现已被证实在植物、真菌、昆虫、果蝇等无脊椎生物中广泛存在[4-6].研究发现,在已分化的哺乳动物细胞中甚至小鼠中也存在抗病毒RNAi作用[7-9].最新研究发现,寨卡病毒感染人类神经前体细胞能够产生大量病毒来源的siRNA,证明人类病毒感染能够有效激发抗病毒RNAi机制[10].
与植物不同,病毒dsRNA同样可以非特异性激活哺乳动物的干扰素(IFN)反应,IFN反应可能会掩盖或抑制宿主的RNAi抗病毒功能[9,11-12].此外,许多病毒还能够编码蛋白抑制宿主的RNAi活动致使不易观察到RNAi现象[13-15].如野田村病毒NoV编码的非结构蛋白B2能够靶向结合双链RNA(dsRNA)抑制Dicer蛋白切割病毒dsRNA或siRNA组装进RISC,从而抑制宿主的抗病毒RNAi功能[16-18],相反,B2蛋白功能缺陷的NoVΔB2感染宿主后能够引起更明显的抗病毒RNAi反应[7-8].
如今,RNAi已成为哺乳动物特异性抗病毒感染的研究热点之一[8,10,12,16].攻毒后病毒基因组拷贝数量、病毒蛋白表达水平或者病毒粒子的装配水平可以用来评价宿主的抗病毒效果,检测病毒来源的siRNA也越来越受到重视[19].20世纪90年代,植物学家在植物中偶然发现RNA沉默现象[20];1998年Fire等人在线虫中发现外源导入的RNA可以抑制线虫肌肉蛋白的表达[21];2013年,我们课题组发现野田村病毒感染哺乳动物细胞后产生大量长度为21~23nt达到可检测水平的病毒特异性siRNA,最近有研究揭示了人类神经前体细胞中的抗病毒RNAi[10],表明抗病毒RNAi在哺乳动物中可能保守存在[7-8].病毒源siRNA是宿主抗病毒RNAi的小分子标志,同时也是宿主防御机制的特异性决定因素[21].RNAi通过病毒基因组同源序列靶向病毒基因组,引导RISC靶向结合病毒基因组,由Dicer蛋白特异性切割病毒双链RNA(dsRNA).病毒源siRNA作为宿主抗病毒RNAi切割病毒基因组的标志性产物,既能反映宿主降解病毒基因组的效率[9,22],还可作为引物合成子代dsRNA,新合成的dsRNA经Dicer切割产生大量的次级siRNA,从而放大RNAi的效应[23].以RNAi通路为靶标设计抗病毒药物,为病毒性疾病的治疗提供了契机.研究表明,当病毒来源小RNA的加工被阻断时,宿主抵抗病毒感染的能力受到极大削弱[9-24].研究指出,针对CSFV非结构蛋白基因构建的siRNA载体可有效抑制CSFV的复制,但抗性细胞中siRNA片段的表达检测制约了RNAi效果的评价[19],因此亟需建立一种简单准确的病毒源siRNA检测方法.
目前检测宿主抗病毒RNAi反应主要依靠小RNA测序技术或放射性同位素标记,但单个样本的小RNA测序费用昂贵,且周期较长,尤其是需要进行大规模样品测序时花费巨大,对经费相对有限的课题组是个不小的挑战.放射性同位素标记实验虽然方法简便,灵敏度高,但容易对人体造成辐射损伤,对安全性要求极高.而国内相关法律法规还不成熟,同时大多数实验室并不具备相关实验条件与资质,可操作性差.因此寻找比较经济的病毒源siRNA检测的可替代方案或在测序前对宿主体内病毒源siRNA进行有效监测与评价具有重要研究价值.
目前已有多种小RNA检测方法,用于检测小干扰RNA(siRNA)或微小RNA(miRNA)等,而选择合适的检测方法是实验成功的关键.Northern blot是小RNA检测的重要方法[25],但特异性小RNA的筛选要求对每种小RNA单独设计探针,步骤繁琐,而且灵敏性有限,样品需求量大,严重制约了小RNA的高通量检测.荧光定量PCR是基因表达定量分析的金标准,尽管siRNA长度不足是一大挑战,基于RT-qPCR仍然开发出多种定量检测方法,比较常用的有Poly(A)加尾法[26]和茎环引物法[27].Poly(A)加尾法适用于高通量筛选小RNA,但特异性较差[26];由于碱基堆积和茎环结构造成的空间约束,茎环引物相较于线性引物更耐高温,特异性和灵敏度也更高[27].TaqMan miRNA矩阵分析可特异性识别成熟的miRNA而不受其前体干扰,准确区分高度同源的小RNA,甚至可精确分辨单个核苷酸的差别[28].Cheng等成功开发出可用于定量检测人工合成的未经修饰或有限修饰的siRNA的操作系统以评估外源siRNA在细胞系或小鼠体内的传递效率与分布特征[27],但仍未有设计茎环状引物在哺乳动物体内或细胞水平特异性检测病毒源siRNA方面的研究.
在前期研究中,我们曾对PR8ΔNS1感染的293T细胞及NoVΔB2感染的乳鼠RNA样品进行小RNA测序,构建了比较完备的病毒源siRNA文库[8,15].在本研究中,我们从PR8ΔNS1病毒来源的siRNA文库[15]中选取2个丰度最高的病毒源siRNA,分别来自NS1片段的正链和负链(16768和3280),同时从NoVΔB2来源的siRNA文库[8]中选取8个siRNAs进行筛选.发现茎环法RT-qPCR检测小鼠病毒源(流感病毒和野田村病毒)siRNA简单方便,易于操作,可以特异性检测病毒源siRNA的丰度,兼具较高的准确性与灵敏度,可以为哺乳动物抗病毒RNAi效价评估提供参考.
1 材料与方法
1.1 材料
人胚肾细胞(293T)为本实验室保存,Balb/c小鼠购买自上海斯莱克实验动物有限公司.RNAi抑制子缺陷的流感病毒(PR8ΔNS1)和野田村病毒(NoVΔB2)为实验室自有[17,29].T载体(pMD20)购买自宝日医生物技术有限公司(Takara中国),大肠杆菌感受态购买自上海唯地生物技术有限公司,DMEM(Gibco)、PBS(Gibco)与TRIzol裂解液(Ambion)购买自Thermo公司,反转录试剂盒购买自Invitrogen公司,荧光定量PCR试剂盒购买自Bio Rad公司.
1.2 细胞培养与病毒感染
293T细胞使用高糖DMEM培养基培养,添加10%胎牛血清(FBS)及1%的青霉素-链霉素双抗,于CO2培养箱37℃培养.
铺板24h后待细胞密度达到80%左右时感染PR8ΔNS1.感染前弃去培养皿中的培养基,用3mL PBS冲洗一遍;弃去PBS后加入3mL不完全培养基(不含FBS),并加入100μL PR8ΔNS1病毒[15];37℃ 培养1h后补加3mL完全培养基(含FBS);细胞培养24h后收细胞.细胞用1mL TRIzol消化并按说明书提取总RNA,以正常未感染细胞作对照.
1.3 动物感染
新生Balb/c小鼠6~8日龄接种NoVΔB2病毒,腹腔注射(i.p.)50μL,病毒滴度为每组7×106个RNA1片段的拷贝[7],对照组接种相同体积的PBS.3dpi取乳鼠后腿肌肉组织样品并提取总RNA.
1.4 内参基因的选择
恰当的标准化方法是基因表达定量分析的关键.本研究参考相关文献确定3个候选内参基因: β-actin,miR-191-3p和miR-103a-3p.其中β-actin是常用内参基因,线性引物扩增产物约200bp.Peltier等发现miR-191-3p和miR-103a-3p在13种正常组织和5对不同的肿瘤/正常邻近组织中的表达高度一致,在统计学上优于常用的参考基因[30],因此作为候选参考基因之一.miR-191-3p和miR-103a-3p仅22bp,因此设计茎环状引物以便于反转录及后续扩增,引物序列见表1.
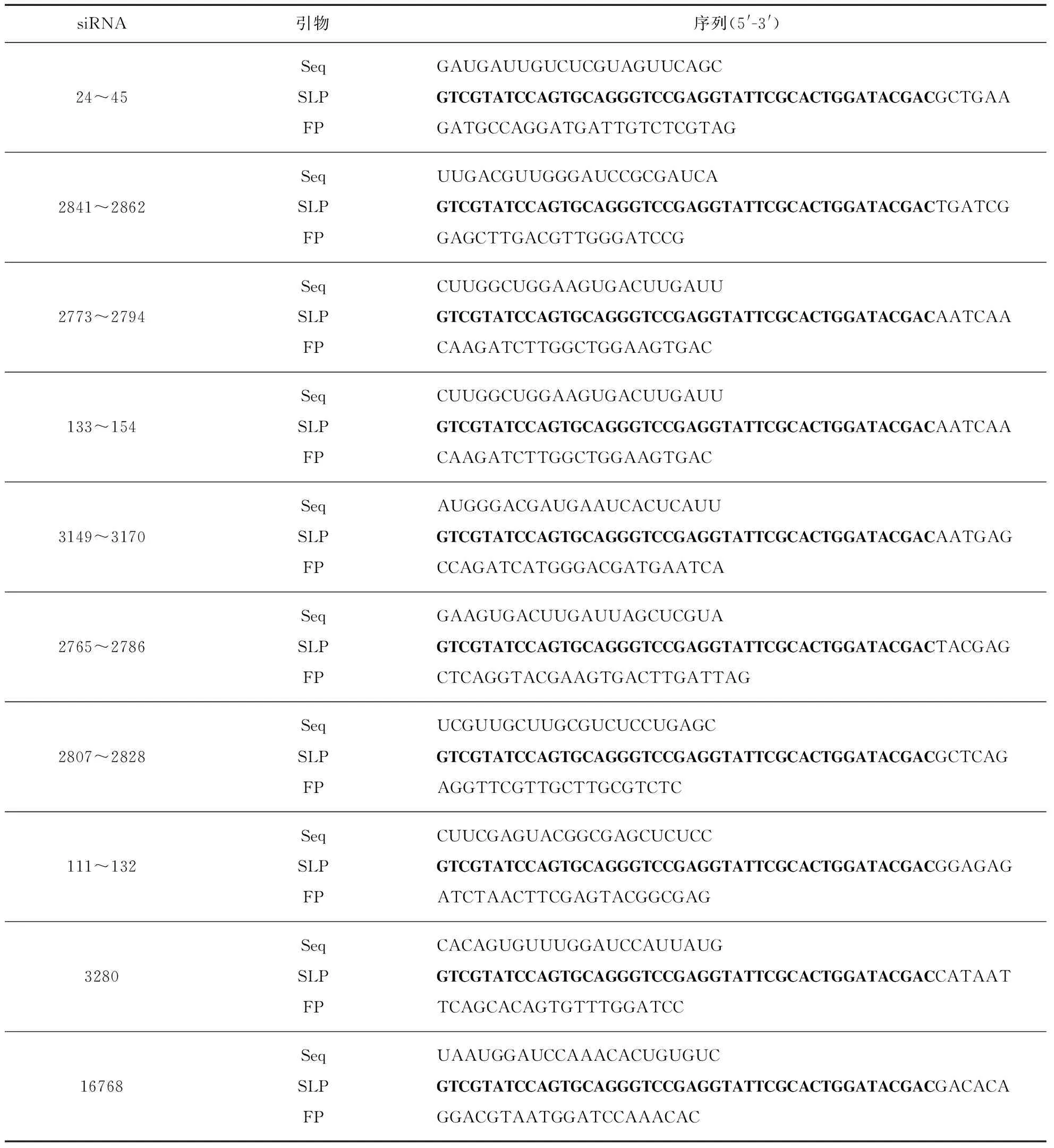
表1 siRNA及引物序列
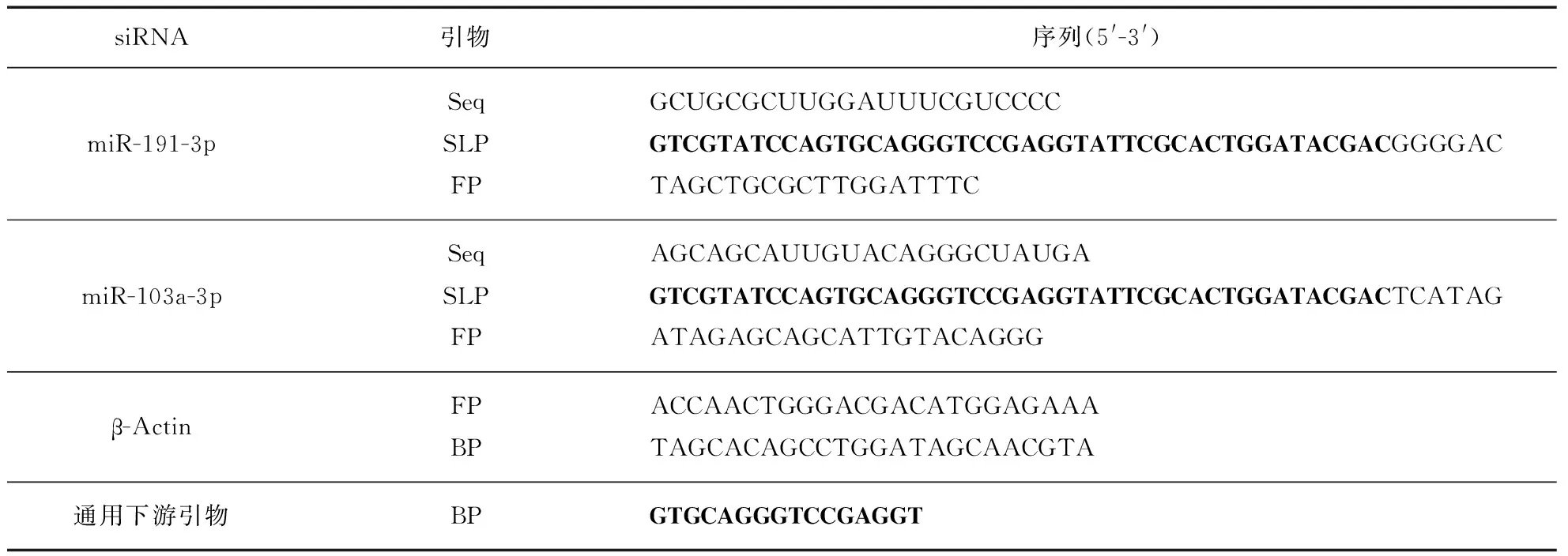
(续表)
1.5 特异性病毒源siRNA的选择
选取293T细胞抗PR8ΔNS1感染产生的表达丰度较高的2个病毒源siRNAs片段(16768和3280)[15],其中16768来自NS1片段的正链,3280来自NS1负链RNA.同样选取Balb/c乳鼠感染NoVΔB2后病毒源siRNA片段,包括不同丰度的小RNA,共8个siRNAs片段[8].
1.6 引物设计与合成
参考Cheng等方法[28]针对PR8ΔNS1和NoVΔB2来源的siRNAs设计引物: 首先,反转录引物由两部分构成,5′端为通用的茎环状结构,序列为(GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTG GATACGAC),3′端由与siRNA反向互补的序列组成,与siRNA互补配对的寡核苷酸数量为6nt(表1).其次,RT-qPCR下游引物为茎环结构上的保守序列,引物序列为GTGCAGGGTCCGAGGT;上游引物分为两部分: 引物3′端为病毒siRNA同源序列,5′端为随机序列(表1),随机序列用于平衡上下游引物退火温度.所用引物均由擎科生物技术有限公司合成.
1.7 茎环法RT-PCR检测病毒来源的siRNAs

图1 茎环法实时荧光定量检测vsiRNA流程图(参考文献[28])Fig.1 Schematic depiction of the protocol for the quantification of vsiRNA by stem-loop primer (Reference from paper[28])
参考文献[28]设计实验方案,病毒来源的siRNAs的检测采取两步法.首先,特异性茎环状引物与病毒源siRNA退火并反转录,然后使用病毒源siRNA特异性的上下游引物扩增逆转录产物(图1).
1.8 总RNA反转录
将提取的293T及乳鼠后腿组织总RNA稀释至1μg/μL,特异性反转录内参基因与病毒源siRNA.
反转录体系为: 1μL茎环引物(2pmol/μL),1μL RNA模板(1μg/μL),1μL dNTPs(10mM),4μL 5×First-strand Buffer,1μL DTT(0.1M),0.5μL反转录酶(200units/μL),用无酶水补至20μL.
反应条件为: 反应体系混合均匀,85℃变性5min,16℃退火30min,50℃反转录30min,70℃ 15min 灭活反转录酶,最后4℃保存.同时每组反转录反应设无反转录酶组作对照,用无酶水补足反应体系.
1.9 荧光定量检测
取上游反转录产物稀释10倍,作为RT-qPCR模板置于冰上.实时荧光定量体系为20μL,其中SYBR Green Supermix(2×)10μL,上下游引物分别0.5μL,无酶水7μL,反转录产物2μL.RT-qPCR每组3个重复,加内参作对照,同时每组设无模板对照组.
1.10 PCR扩增目的基因,载体构建与转化
PCR反应体系为: Takara Ex Taq酶0.25μL,10×Ex Taq Buffer 5μL,4μL dNTPs(10mmol/L),上下游引物各2μL,反转录产物100ng,用无酶水补足至50μL.
反应条件为: 反应体系混合均匀后98℃变性10s,55℃退火30s,72℃延伸1min,36个循环.PCR扩增产物核酸胶电泳鉴定,目的片段连接T载体,转化E.coliDH5α感受态细胞.感受态细胞经涂板、过夜培养后挑单克隆,扩大培养后提取质粒、送测序.
2 结果与分析

表2 RNA质量检测
2.1 RNA质量检测
收取PR8ΔNS1感染24h和未感染的293T细胞,NoVΔB2感染和未感染组Balb/c乳鼠第3天收取乳鼠后腿肌肉组织,利用TRIzol法抽提细胞RNA并检测RNA质量.检测结果表明,病毒感染组与对照组所提取得RNA样品纯度较高,A260/A280在2.0左右,表明蛋白污染较少;部分样品A260/A230较小(293T mock),表明该样品RNA中可能含有少量有机试剂残留.
2.2 内参基因的选择
以提取的总RNA为模板,加入特异性茎环状引物反转录候选内参基因,取稀释10倍后的反转录产物进行RT-qPCR,结果如图2所示.
PR8ΔNS1感染293T组miR-191-3p的Ct值为31.5左右,与对照组Ct值差异极显著(22.2),因此不适合作为内参基因;而β-Actin的Ct值为22.5左右,与对照组ΔCt为2.1;miR-103a-3p的Ct值为24.5,ΔCt小于1(图2(a)).考虑到miR-103a-3p长22nt,与病毒源siRNA的特征更加相似,因此选择miR-103a-3p作为内参基因检测PR8ΔNS1来源的siRNA.
NoVΔB2感染乳鼠组miR-191-3p,miR-103a-3p和β-Actin的绝对Ct值分别为24.7,25.9和23.5左右.感染组与对照组相比,miR-191-3p的Ct值无显著性差异(P=0.43);miR-103a-3p和β-Actin的Ct值虽然差异显著,但ΔCt均在1左右(图2(b)).本研究优先选择miR-191-3p作为内参基因.

图2 内参基因荧光定量结果Fig.2 RT-qPCR results of reference genes
2.3 荧光定量检测病毒源siRNA
PR8ΔNS1感染293T细胞24h后提取RNA,以miR-103a-3p为内参基因,检测病毒源siRNA,结果如图3所示.与对照组相比,PR8ΔNS1感染组内参基因miR-103a-3p ΔCt为0.7;病毒源siRNA的Ct值与表达水平有极显著性差异(图3(a)).其中16768的表达水平是对照组的800倍左右,3280表达量是对照组的700倍(图3(b)),因此16768和3280均可以作为目标基因评价宿主抗病毒RNAi效率.
NoVΔB2感染Balb/c乳鼠3天后收样提取RNA,以miR-191-3p为内参基因,检测病毒源siRNA,结果如图4所示.与对照组相比,NoVΔB2感染组病毒源siRNA 24~45,2841~2862,2807~2828和111~132的绝对Ct值有显著性差异(图4(a)),表达水平经内参基因标准化后24~45有显著性差异(图4(b)),因此将24~45作为目标基因评价乳鼠抗病毒RNAi的效率.
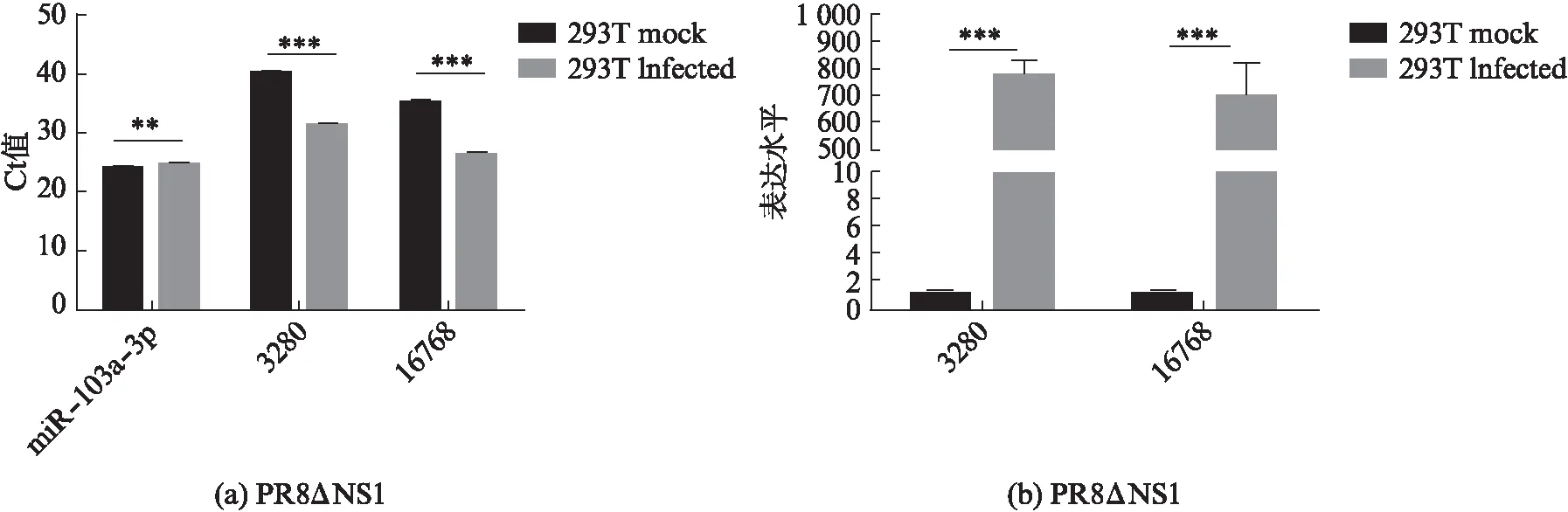
图3 病毒源siRNA荧光定量结果Fig.3 RT-qPCR results of virus-derived siRNA

图4 病毒源siRNA荧光定量结果Fig.4 RT-qPCR results of virus-derived siRNA
2.4 RT-qPCR重复数据的一致性
荧光定量PCR每组3个重复(部分数据,表3),293T和Balb/c乳鼠的感染组与空白对照组组内偏倚较小,所有数据的方差均小于0.5,大多数在0.0至0.2之间,表明数据重复性较好,结果一致,该方法稳定可靠.293T对照组病毒源特异性小干扰RNA片段3280和16768的平均Ct值分别为40.0和35.1,远大于感染组Ct值(分别为31.1和26.4),说明检测背景低,可通过Ct值的差异有效区分阴性和阳性结果,具有很高的特异性.

表3 荧光定量PCR重复数据一致性比较
2.5 PCR扩增病毒源siRNAs
病毒源siRNAs扩增产物经凝胶电泳检测并测序,结果如图5所示.未加模板的反转录产物及PCR扩增产物无明显条带,表明引物未形成引物二聚体或多聚体;反转录时不加反转录酶,荧光定量PCR不能扩增出有效条带,说明RNA样品中无DNA污染,正常实验组条带明亮且单一,无杂带,引物特异性高(图5(a)).PCR产物测序结果表明扩增序列与PR8ΔNS1源的siRNA序列完全一致(图5(b)),该方法可以用来快速鉴定宿主体内的病毒源的siRNA,且具有较高的准确性.
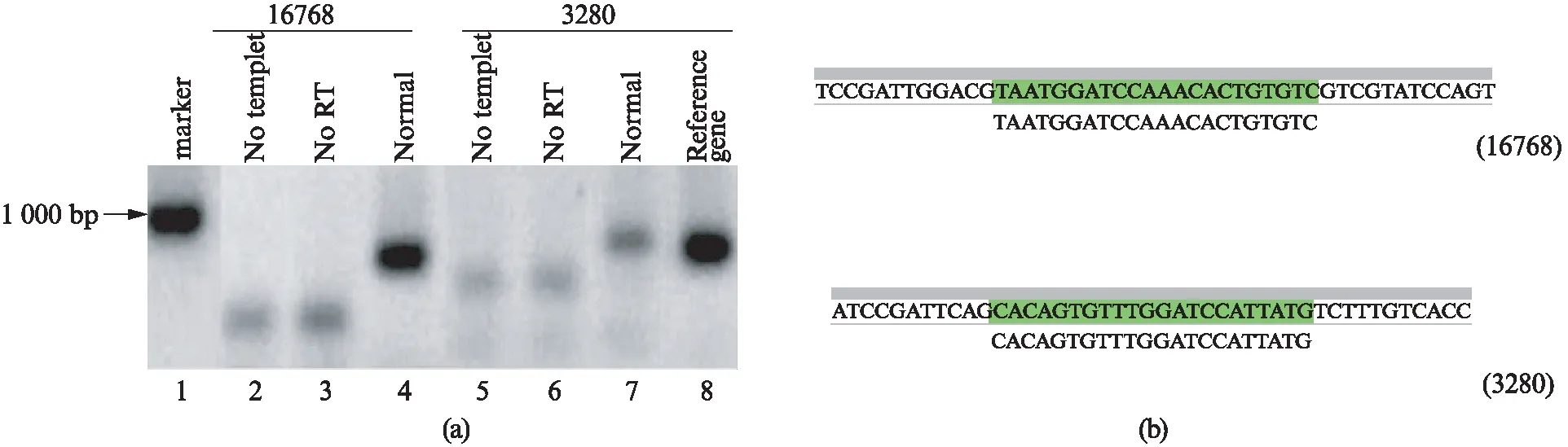
图5 PCR扩增293T抗PR8ΔNS1 vsiRNAsFig.5 PCR amplify 293T anti-PR8ΔNS1 vsiRNAs
2.6 病毒源siRNA RT-PCR结果分析
PR8ΔNS1感染组病毒源siRNA 16768和3280绝对Ct值分别为26.4和31.1,即16768基因表达量是3280的32倍(图3(a));与对照组相比,16768和3280相对Ct值均为9,根据内参标准化后,基因表达量亦接近1∶1(图3(b)).而病毒源siRNA文库数据则显示,16768的丰度是3280的5倍[15],与RT-PCR结果有一定差异.
同样,NoVΔB2感染组病毒源siRNAs 24~45,2841~2862,2807~2828和111~132的绝对Ct值分别为32.0,33.4,34.7,36.2(图4(a));与对照组相比,相对Ct值分别为8,3,2,2,基因表达量比值接近于50∶1∶1∶1(图4(b)).而NoVΔB2的siRNA文库数据则显示,24~45,2841~2862,2807~2828和111~132的丰度比值接近于160∶40∶1∶1[8],虽然RT-PCR与小RNA测序结果中病毒源siRNAs丰度存在差异,但总趋势一致,即高丰度的siRNA标准化后的表达量也比较高.
3 讨 论
RNA病毒感染植物、昆虫、线虫、哺乳动物等均能激活宿主的RNAi抗病毒功能,宿主识别病毒dsRNA并将其切割成siRNA[31].RNAi发生于除原核生物以外的所有真核生物细胞内.RNAi的效率非常高,少量的双链RNA分子通过宿主的信号级联放大效应就能引起系统性的RNA干扰,抑制相应基因的表达[23].研究发现,dsRNA可以越过细胞间的屏障,在不同细胞间远距离传递并维持信号,甚至传播至整个有机体[32].RNA病毒感染后提取总RNA进行小RNA测序,分析RNA样品中的小RNA含量、大小及分布特征是否与RNAi产物特征一致,以判断病毒是否激活了宿主的抗病毒RNAi功能.Northern blot是检测小RNA片段的重要方法,但哺乳动物切割Nov或流感病毒产生的小RNA片段reads总数较低.通常,低丰度的小RNA在克隆、RNA印记或阵列分析中往往检测不到,检测结果难以排除假阴性,同时不利于开展大批量小RNA片段的筛选工作.而更为灵敏的放射性同位素标记试验在国内尚未普及,大多数实验室也不具备操作条件(本课题组曾在国外应用该方法成功检测到病毒源小RNA).小RNA测序结果准确,但费用高,周期长,因此选择siRNA检测的可替代方案具有重要研究价值.
RT-qPCR是基因表达定量的重要标准[33-34],但如何设计实验方案检测小RNA是个重大难题.传统的荧光定量引物对RNA片段长度有一定要求,不能扩增长度过短的RNA片段,如siRNA,miRNA,发卡RNA(shRNA),与PIWI相互作用的RNA(piRNA),重复相关小干扰RNA(rasiRNA)等.而茎环状引物通过反转录在小RNA的基础上添加了数十个碱基从而解决了病毒源siRNA长度过短的问题.茎环状引物的碱基堆积可以有效改善引物的热稳定性,特异性和灵敏度也更高.
恰当的标准化方法是基因表达定量分析的关键,但常常被忽视.本研究参考相关文献确定3个候选内参基因,其中β-actin是常用内参基因,miR-191-3p和miR-103a-3p则在哺乳动物多种组织或细胞系中稳定表达[30].我们发现293T细胞感染PR8ΔNS1对miR-103a-3p产生的影响基本可控,ΔCt值仅0.67,甚至略优于β-actin,因此选择miR-103a-3p作为内参基因检测PR8ΔNS1感染293T产生的siRNA.NoVΔB2感染Balb/c乳鼠后miR-191-3p的表达未产生显著性变化(P=0.43),因此我们选择miR-191-3p作为内参基因检测NoVΔB2感染Balb/c乳鼠后产生的siRNA.
目前已发表的相似的研究中,大多通过体外转录制备病毒源siRNA标准品,绘制标准曲线,建立定量标准曲线Ct值与模板拷贝数对数之间的线性关系,筛选兼具特异性和灵敏度的siRNA[18].本研究采用相对定量技术,通过将病毒RNA样品梯度稀释至1000倍,建立了病毒拷贝数对数与Ct值之间良好的线性关系,相关系数R2为0.9979,扩增效率为95%(数据未展示),表明该系统稳定性高,结果可信.
本实验针对流感病毒和野田村病毒来源的不同的siRNAs设计多组茎环状引物及上下游引物,检测病毒源siRNAs,成功筛选出能够显著区分病毒感染组与对照组的siRNAs,可以作为靶基因验证宿主抗病毒RNAi功能的启动.由于siRNA和miRNA长度均为21nt左右,且细胞内源性有高达数百种高表达的miRNA,因此,排除宿主细胞对检测结果的影响非常必要.在本研究中,所有荧光定量PCR试验均进行3次重复,重复样品病毒源小RNA的Ct值差异不显著,ΔCt值趋向一致,偏倚较小.而对照组病毒源特异性小干扰RNA片段均超过35,远大于感染组vsiRNA的Ct值,表明该体系非特异性扩增低,受小鼠或293T细胞内源性miRNA的干扰较小,具有较高的检测特异性,可通过Ct值的差异有效区分病毒来源的特异性siRNA.Chiang等[27]应用茎环法设计了多达100个siRNA检测体系,结果94.4%的阴性对照组Ct值在35以上,Ct值在30~35之间的不到5%,而阳性结果均小于30.可见该体系中细胞miRNA造成的背景影响非常小,并不会干扰目的基因的检测.293T mock组A260/A230略低于其他组,但实时荧光定量PCR溶解曲线提示只有单一峰,表明该样品质量不会影响荧光定量的检测.PR8ΔNS1感染293T时,病毒源siRNA 16768和3280基因表达量接近1∶1(图3(b)),与siRNA测序文库中的检测量有一定差异(5∶ 1)[13].这可能是由不同的检测方法造成的系统误差.小RNA测序基于第二代测序技术,易受多种因素影响导致测序结果出现偏差.反转录时所用的随机引物可能会引起前几位核苷酸的组成具有一定的偏好性;通常序列前端碱基错误率较高,而随着测序序列的延伸,序列末端的错误率也大大提升.同样,经内参标准化后NoVΔB2感染组病毒源siRNAs中的24~45,2841~2862,2807~2828和111~132的基因表达量比值接近于50∶1∶1∶1(图4(b)),虽然与小RNA测序结果中病毒源siRNA丰度有一定差异(160∶ 40∶1∶1)[8],但趋势一致,即高丰度的siRNA标准化后的表达量高于低丰度siRNA.测序结果也证实茎环法RT-qPCR扩增序列与病毒源siRNA序列完全一致(数据未展示),Cheng等[28]采用Northern blot与基于放射性同位素标记的探针法RT-qPCR检测miRNA结果做对比.结果,基于Northern blot的miRNA分析重复性低且一致性差,小鼠组织不同miRNA荧光信号强度与Ct值相关系数有较大波动(0.751~0.916).此外,由于工作原理的限制,导致Northern blot无法准确区分同源序列.然而,TaqMan miRNA矩阵分析却可以特异性识别成熟的miRNA而不受其前体干扰,可以准确区分高度同源的小RNA,甚至精确分辨单个核苷酸的差别,显示出该方案优异的特异性.在本研究中,我们通过设计茎环引物特异性扩增病毒源小RNA取得了较好结果.该系统可以检测低丰度的siRNA,siRNA序列经测序验证无误.RT-qPCR结果与小RNA序列文库中病毒源siRNA丰度进行比较,两种检测结果显示高丰度的siRNA标准化后的表达量高于低丰度siRNA,而丰度值在同一级别的siRNA表达量亦接近1∶1.近年来,有研究[35]针对登革病毒3′端非结构蛋白编码区和基孔肯雅病毒5′端非结构蛋白编码区设计引物和探针,应用实时荧光定量PCR技术建立定量标准曲线Ct值与病毒拷贝数的对数值之间的线性关系,并将其应用至临床检测300多份血清标本中病毒来源的RNA片段.结果显示,用该方法检测登革热患者血清,灵敏度达到100%,特异度89.3%,阳性和阴性预测值分别为92.1%和100%;基孔肯雅热患者的灵敏度、特异性及阴、阳性预测值均达到100%,为早期筛查、鉴别病毒感染提供了重要依据[35].这一研究与本文研究思路有异曲同工之处.在我们的研究系统中,通过参考病毒siRNA文库的丰度,病毒特异性RNA片段的选择更加科学,针对性和特异性更强,表明荧光定量检测技术不仅简单易操作,同时具有较高的特异性和准确性.但该系统是否可以直接应用到其他细胞系或哺乳动物组织中还需要更多的研究结果支持.
猜你喜欢
科学(2020年3期)2020-11-26 08:18:22
当代水产(2020年3期)2020-06-15 12:03:02
传媒评论(2019年12期)2019-08-24 07:55:10
传媒评论(2017年3期)2017-06-13 09:18:10
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:42
新闻传播(2015年10期)2015-07-18 11:05:39
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
