
金属元素掺杂硫化钼催化加氢研究进展
2019-10-11 03:06:50周伟霞楚素娅张陈洋刘勇军
石油化工 2019年9期
周伟霞,郑 毅,楚素娅,张陈洋,刘勇军
(华侨大学 化工学院,福建 厦门 361021)
加氢处理是石油炼制生产清洁油品的重要工艺之一,它可通过加氢的方式脱除原料中的硫化物、氮化物和芳烃等[1-4]。硫化钼系材料是典型的加氢处理催化剂,尤其是加氢脱硫(HDS),最常见的活性组分组合为CoMo,NiMo,NiW。Co(Ni)MoW 三元金属硫化物也成为重要的加氢处理催化剂。由于对活性相结构和催化剂作用机理认识水平的不断提高,硫化钼系材料的催化加氢活性持续提高。但目前对活性中心的微观结构及电子特性、助剂作用的本质及含硫、氮等分子的吸附机理等问题限制了硫化钼系材料HDS 活性的进一步提高。近年来,由于TEM、扫描隧道显微镜(STM)等表征方法与基于密度泛函理论(DFT)计算工具的发展,加深了人们对催化剂颗粒的结构和活性位点性质的了解[5-7]。目前,研究者已成功应用水热/溶剂热法、氧化前体硫化法和热分解法等方法制备了不同加氢活性的硫化钼基催化剂。
本文主要从催化剂活性中心结构及吸附机理两个方面阐述了钴、镍助剂促进作用的研究进展,对硫化钼系催化剂的未来研究方向进行了展望。
1 制备方法
1.1 水热/溶剂热法
水热法是在高温高压的密闭反应器中,使在常压条件下不易溶于水的物质溶解,继而发生化学反应,产物过饱和析出的方法[8],具有反应条件温和、产物形貌可控的优点。通过调控水热条件(温度、压力、溶剂等)、有机助剂可以制备不同形貌(片状、花状、球状等)的硫化钼基催化剂。Akram 等[9]以乙二胺为溶剂,制备了CoMoS2和NiMoS2纳米球,直径在450~1 000 nm 之间。独特的花状纳米结构易形成高密度的活性位点。Lai 等[10]以元素硫和(NH4)2S 为硫源分别制备了3D 纳米花状和片状NiMoS 催化剂,花状MoS2团簇出现大量的位错和扭曲,使4,6-二甲基二苯并噻吩(4,6-DMDBT)的HDS 转化率高达95%,甲基环己基甲苯选择性为70%以上。水热温度与压力会影响金属掺杂硫化钼的团簇结构,从而改变催化加氢活性与选择性。Yoosuk 等[11]研究发现,水热温度升高导致MoS2板坯的曲率增加而长度减小,4,6-DMDBT 转化率从41.6%增加至51.9%,加氢反应(HYD)/直接脱硫(DDS)比率从1.5 增加到2.9;而高压也促进4,6-DMDBT 的转化,但对HYD 路径稍有抑制。Wang 等[12]还发现,经高温水热的CoMoS 更有利于对甲苯酚的转化和直接脱氧路径,而高压促进对甲苯酚转化和氢化脱水反应;对甲苯酚的高转化率(100%)归因于CoMoS 催化剂更短的板长度和更高的堆叠数。通常仅用水或有机溶剂制备的硫化钼难以定性、稳定性差,一般通过加入助剂或与其他方法相结合来稳定硫原子,降低氧化程度。Wang 等[13]利用聚乙烯吡咯烷酮(PVP)辅助水热法合成碳涂覆的CoS2-MoS2催化剂,疏水性包覆碳抑制了催化剂中硫的损失,直接脱氧活性高达96%。助剂钴、镍的引入会阻碍硫化钼颗粒的生长,改善结构特征。Yoosuk[14]等经一步水热法制备了无定形NiMoS 催化剂,实验结果表明,镍的加入显著促进了加氢脱氧活性,苯酚转化率达到96.2 %,归因于镍促进硫化钼团簇中大量Brim 位点的存在。综上所述,采用水热/溶剂热法合成MoS2系催化剂,可根据前体浓度、反应温度、压力和反应时间等条件控制产物的形貌,但存在产量少、晶型较差和反应时间长等不足。
1.2 氧化前体硫化法
对前体氧化物进行硫化是合成MoS2基催化剂的常用途径,一般以H2S/H2气氛为硫源和还原剂。该方法能够通过调控前体中的不同金属比、添加有机助剂等,制备不同结构的氧化物前体,但硫化过程通常不充分,高温易使产物团聚。Liu 等[15]通过调节PVP 添加量合成了α-NiMoO4,β-NiMoO4两种相态的氧化物前体,并发现β 相NiMoS 催化剂的二苯并噻吩(DBT)转化率比α 相硫化物高一倍以上。这是因为β 相前体中的氧化物更易还原,并且它可能更有利于NiMoS 活性相的形成。前体的类型是制备和优化HDS 反应活性材料的基础,体系中不同的活性组分及组分元素的不同摩尔比大大影响了催化剂HDS 活性和选择性。Amaya 等[16]比较了由层状氧化前体硫化获得的CoMoW,NiMoW硫化物的HDS 活性,DBT 转化率分别为25%和93%,两者的活性差异与镍材料的弯曲形态和较高氢化能力相关。Liu 等[17]制备了具有不同Ni/Zn摩尔比的NiZnMo 硫化物,其中Ni9.5Zn0.5Mo10的DBT 转化率最高;而Zn10Mo10的苯基环己烷选择性最高可超过80%。作为“氧化物路线”的替代方案,从合成溶液中直接沉淀硫化物有利于材料的活性,这通常归因于改善的Ni(Co)Mo 相互作用[18]。Genuit 等[19]以镍或钴盐及硫代钼酸盐为原料制备了镍、钴掺杂的硫化钼催化剂,脱硫率比商业负载体系高六倍。一般大分子硫化物在MoS2活性组分的角位被吸附脱除[20],因此催化剂的脱硫活性和选择性与MoS2团簇结构紧密相关。曾鹤等[21]比较了共沉淀法和水热法制备的NiMoW 催化剂对催化裂化柴油的HDS 反应性能,发现前者的HDS率高达98.8%,硫化态催化剂的平均板长和堆叠数要大于水热法。Jeong 等[22]提出了一种新的核-壳纳米团簇氧化物的合成方法,经过高温煅烧后,也能够对NiMoS2纳米粒子典型的板坯结构(板坯长度、堆叠厚度)进行调控。
1.3 热分解法
MoS2系催化剂也可以由高温加热分解前体的方法来制备,其中前体既可以是单源分子前体(单个反应分子含有产物需要的所有元素),又可以是非单源分子前体(由多个反应物分子提供最终产物的所有元素)。Guo 等[23]通过热分解法成功制备了具有超小尺寸的单层MoS2,MoNiS,MoCoS。实验结果表明,噻吩的转化率分别为64%,85%,84%;吡啶的共存对噻吩HDS 反应有一定的抑制效应。王岩[24]由单源分子前体热解合成了MoS2和NiMoS 催化剂,DBT 的转化率分别为63%和97%,同时镍的掺杂也大大改善了催化剂的耐氮性能。用热分解法制备的硫化物结晶性较好,但前体热解过程涉及有毒物质硫化铵或硫化氢,暂时不能大规模生产。
2 金属掺杂MoS2促进作用机理
2.1 结构效应
人们普遍认为活性位点位于催化剂颗粒的边缘,可能涉及边缘(金属和硫边缘)、角位和Brim 位点[25]。图1 为NiMoS 和CoMoS 的代表性粒子模型。由图1 可知,边缘和角位属于配位不饱和(CUS)位点,不饱和的Mo 原子具有未被占据的d态高费米能级,这就允许它与给电子分子的强结合;另一方面,Brim 位点不需要不饱和度且具有金属特性,这是由于它存在跨越费米能级的一维边缘状态[26]。硫化钼系催化剂不同位点的性质差异直接影响了催化加氢活性。Šarić 等[27]比较了含硫分子在边缘、角位和基面的吸附自由能及硫空位形成能,解释了边缘和角位对HDS 反应的活性,而基面是惰性的;并提出整个S 边缘是具有活性的,但对于Mo 边缘,仅空位具有活性。

图1 NiMoS(a)和CoMoS(b)的代表性粒子模型Fig.1 Representative particle models for NiMoS(a) and CoMoS(b).
助剂的引入和制备条件影响催化剂颗粒的边缘及其活性位点的性质。Walton 等[28]通过STM 表征观察到MoS2团簇为三角形,以硫二聚体(100%S 覆盖度)饱和的Mo 边缘终止;而CoMoS2纳米颗粒表现为截短的六边形,由Co 促进的S 边(50%S 覆盖度)和Mo 边组成。促进作用归因于50% S覆盖度的S 边缘处更强的相互作用和在边缘S—H基团中有益的氢结合能。Grønborg 等[29]研究了还原条件对(Co)MoS2的团簇结构和边缘S 覆盖度的影响。实验结果表明,s-MoS2(H2S)和r-MoS2(H2)团簇分别为三角形和截断的六边形,Mo 边覆盖度由100% S 降低为50% S。对于CoMoS,H2还原未出现结构重构,但所有边缘覆盖度都降低至50% S;这证明了在Co 促进的边缘上存在催化相关的S—H 基团。Lauritsen 等[30]也在STM 表征中观察到Ni 在MoS2边缘上可部分/完全取代Mo 原子。促进效应的性质本质上是电子性的,这是由于Co 对Mo 的电子给予弱化了金属—硫键强度,使其能够达到与优化的HDS 性能相对应的值[31]。尽管CoMoS 相在文献中已有很好的描述,但Co9S8和MoS2实际共存区域的协同效应尚不完全清楚。Ramos 等[32]模拟了Co9S8/MoS2的界面模型(见图2),并且合理描述了两个体相之间的表面接触区域,界面Mo 原子变为五重配位(体相MoS2为六重配位),表明存在潜在的空位;DFT 计算结果表明,从Co 到Mo 的强电子供给使Mo—S 键的强度减弱,使其易形成CUS 位点。
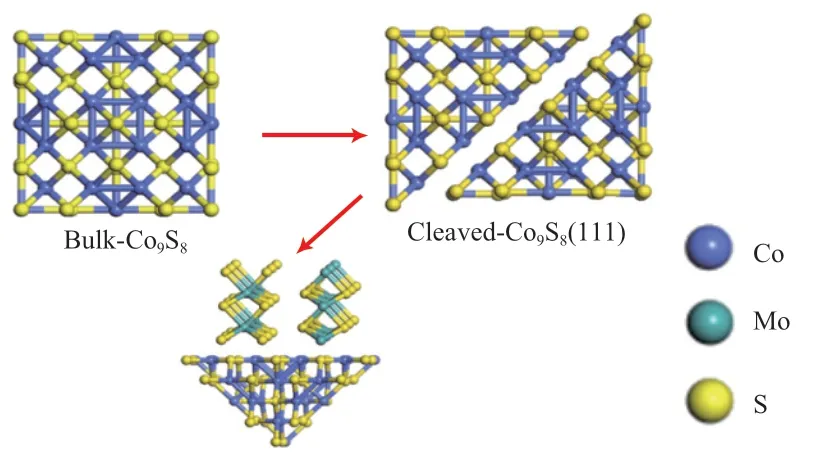
图2 模型界面建立过程Fig.2 Model interface establishment process.
2.2 吸附效应
在HDS 催化反应中,除去具有空间位阻的含硫分子(如4-DMDBT,4,6-DMDBT 等)仍然是主要的挑战,由于该分子在空间上被甲基官能团屏蔽[33]。为了提高催化活性,需要对这类分子与催化剂之间的相互作用进行深入的理解。
不同结构的分子以不同的吸附构型吸附于暴露的CUS 位点或Brim 位点,4,6-DMDBT 类大分子主要通过离域π模式吸附于Brim 位点进行HYD 反应。Grønborg 等[34]联合STM 和DFT 研究了4,6-DMDBT 在CoMoS 相催化剂上的吸附构型。研究表明,离域π模式是4,6-DMDBT 进行HYD 路径的吸附方式,以Brim 构型进行物理吸附;而DDS 路径中该分子以σ模式吸附,并且仅能吸附在CoMoS 团簇的S 边-角空位。šarić 等[35]进一步研究了含硫分子在CoMoS 的边缘、角位和基面上的吸附(见图3)。实验结果表明,4,6-DMDBT倾向于以Brim 构型吸附在S 边缘,它导致更强的相互作用,从而通过HYD 路径脱硫。而小分子(甲硫醇、噻吩、DBT 等)以边缘构型通过化学吸附优选Mo 边或角位的空位,遵循DDS 路径。硫空位通常被认为不可直接吸附完好的烷基取代DBT类分子,钴镍掺杂的硫化钼团簇能够为含硫分子提供更多的吸附位点,特别是团簇边缘Brim 位点能够大大减弱4,6-DMDBT 类分子的位阻效应,促进了在CUS 位点的有效吸附。研究提出了具有空间位阻的大分子的脱除涉及氢化和脱硫两个步骤,氢化步骤使4,6-DMDBT 的苯环饱和,这会增加甲基取代基键的灵活性,使分子的空间位阻减弱,促进分子在Brim 位点的吸附;同时S 空位具有较高的硫醇盐形成驱动力,能够使氢化分子强烈吸附在S 空位上来脱除硫。
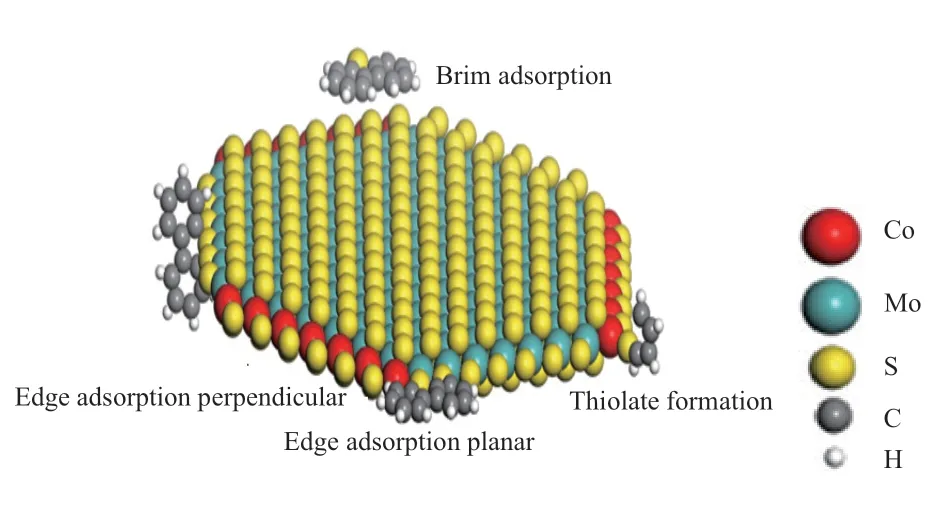
图3 理想CoMoS 颗粒的模型及含硫分子的不同吸附构型Fig.3 Model of ideal CoMoS particles and different adsorption configurations of sulfur-containing molecules.
在工业条件下,有机氮化合物的存在会影响含硫分子的吸附,从而抑制HDS 反应[36]。Rangarajan 等[37]运用DFT 研究了20 种含氮/硫化合物在CoMoS 催化剂S 边缘的吸附。根据结合能显示,含氮化合物比含硫分子更强地吸附在Brim位点,导致4,6-DMDBT 类的HYD 反应受到强烈抑制;而噻吩类小分子比氮化物更强地吸附在CUS 位点。Salazar 等[38]采用STM 表征更直观地探究了吡啶和喹啉的吸附构型和扩散特征,进一步证实了含氮化合物与氢化反应中相同活性边缘位点上含硫分子的竞争吸附。
综上所述,钴、镍助剂的加入改变了MoS2团簇的微观结构,暴露更多的Brim 位点和CUS 位点,分别作为甲基取代DBT 类分子的氢化步骤和脱硫步骤的强吸附位点。另一方面,反应物中氮化物与含硫化合物在氢化反应活性位点上存在较强的竞争吸附,抑制了大分子硫化物的脱除。
3 结语
STM 表征对原子缺陷、边缘位点和角位很敏感;通过加入相关反应物,可观察反应中间体并通过此方式追踪反应路径用于研究活性位点的性质。因此,需要开发具有足够真实性能的模型系统,使它们与工业催化剂中存在的Co(Ni)MoS2纳米颗粒相当。真实的汽油原料会含有抑制HDS 反应的含氮化合物,这需要加深含氮化合物对HDS 反应的抑制机理的研究,指导开发具有高HDS 反应活性和使含氮化合物含量最小的抑制催化剂。根据硫化钼基材料的“结构-性能”机制,对现有的制备方法进行改进或开发新的方法,从而实现对产物形貌、结构和性能的精准控制。
猜你喜欢
天津医科大学学报(2021年1期)2021-12-05 11:11:05
西南石油大学学报(自然科学版)(2021年3期)2021-07-16 05:27:18
环境保护与循环经济(2021年12期)2021-03-16 05:51:12
中国特种设备安全(2019年3期)2019-04-22 05:05:38
山东工业技术(2016年15期)2016-12-01 05:30:43
现代检验医学杂志(2016年5期)2016-08-20 03:17:08
石油知识(2016年2期)2016-02-28 16:19:49
环境科技(2015年5期)2015-11-08 12:08:58
河南科技(2014年15期)2014-02-27 14:12:38
茶叶通讯(2014年2期)2014-02-27 07:55:40
