
TNFα inhibitor C87 sensitizes EGFRvIII transfected glioblastoma cells to gefitinib by a concurrent blockade of TNFα signaling
2019-09-29 06:27:50LiMaChunhuaSheQianShiQiangYinXinxinJiYongrongWangYulongFanXinyaoKongPengLiZengfengSunXiaohuiZhangZhenZhangJianWangTongWangYuanfuXuWenliangLi
Cancer Biology & Medicine 2019年3期
Li Ma, Chunhua She, Qian Shi, Qiang Yin, Xinxin Ji, Yongrong Wang, Yulong Fan, Xinyao Kong, Peng Li,Zengfeng Sun, Xiaohui Zhang, Zhen Zhang, Jian Wang, Tong Wang, Yuanfu Xu, Wenliang Li
1Department of Neuro-Oncology and Neurosurgery, Tianjin Medical University Cancer Institute & Hospital, National Clinical Research Center for Cancer; Key Laboratory of Cancer Prevention and Therapy, Tianjin; Key Laboratory of Cancer Immunology and Biotherapy, Tianjin's Clinical Research Center for Cancer, Tianjin 300060, China; 2State Key Laboratory of Experimental Hematology, CAMS Key Laboratory for Prevention and Control of Hematological Disease Treatment Related Infection, National Clinical Research Center for Hematological Disorders, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300020, China; 3Department of Immunology, Tianjin Medical University Cancer Institute & Hospital, National Clinical Research Center for Cancer; Key Laboratory of Cancer Prevention and Therapy, Tianjin; Key Laboratory of Cancer Immunology and Biotherapy, Tianjin’s Clinical Research Center for Cancer, Tianjin 300060, China
ABSTRACT Objective: More than half of human glioblastomas show EGFR gene amplification and mutation, but EGFR inhibitors have not been effective in treating EGFR-positive glioblastoma patients. The mechanism behind this type of primary resistance is not well understood. The aim of this study was to investigate gefitinib resistance in glioblastoma, and explore ways to circumvent this significant clinical problem.Methods: MTT method was used to test the cell viability after EGFR-positive glioblastoma cells were treated with indicated drugs;real-time quantitative PCR method was included to detect the TNFα mRNA levels in glioma tissues and cell lines. ELISA was introduced to measure the TNFα protein levels in cell culture supernatant of glioblastoma cells treated with gefitinib. Western blot was used to detect the activity change of intracellular kinases in drug-treated glioblastoma cells. Two mouse xenograft tumor models were carried out to evaluate the in vivo effects of a combination of EGFR and TNFα inhibitors.Results: We found that glioblastoma resistance to gefitinib may be mediated by an adaptive pro-survival TNFα-JNK-Axl signaling axis, and that high TNFα levels in the glioblastoma microenvironment may further intensify primary resistance. A combination of the TNFα-specific small-molecule inhibitor C87 and gefitinib significantly enhanced the sensitivity of glioblastoma cells to gefitinib in vitro and in vivo.Conclusions: Our findings provide a possible explanation for the primary resistance of glioblastoma to EGFR inhibitors and suggest that dual blockade of TNFα and EGFR may be a viable therapeutic strategy for the treatment of patients with chemotherapy-refractory advanced glioblastoma.
KEYWORDS Glioblastoma; EGFR; TNFα; inhibitor; drug resistance
Introduction
Glioma is the most common primary tumor of the central nervous system, and is characterized by aggressive tumors that invade the normal brain parenchyma1. Complete resection of gliomas is difficult because these tumors are not encapsulated and are typically enmeshed with other brain cells2. Gliomas are associated with poor prognosis as chemotherapies, and anti-tumor traditional Chinese medicines are hampered by factors such as the inability to cross the blood-brain barrier3. Glioblastoma accounts for more than 60% of all newly diagnosed glioma cases, and is categorized by the World Health Organization (WHO) as grade IV glioma. Glioblastomas are more malignant than lower grade gliomas (WHO II-III), such as astrocytomas and oligodendrogliomas3-5. The standard treatment for newly diagnosed glioblastoma consists of surgical resection,radiotherapy, and concomitant adjuvant chemotherapy with temozolomide (TMZ). Despite this treatment approach, the median overall survival of glioblastoma patients is only 14.6 months, with a 2-year survival rate of less than 26.5%and a 5-year survival rate of less than 5%6.
Precision medicine and targeted therapies specifically target oncogenes or closely related signaling pathways to inhibit tumor growth. In recent years, targeted therapies have achieved significant clinical efficacy and have become a standard treatment option along with surgery, radiotherapy,and chemotherapy7. Most notably, the discovery of theEGFRoncogene and the development of EGFR-targeting tyrosine kinase inhibitors (TKIs) are important milestones in the development of tumor-targeted therapy8,9. Furthermore,small-molecule inhibitors targeting EGFR, such as gefitinib,erlotinib, icotinib, and lapatinib, have been shown to be beneficial for numerous patients with advanced non-small cell lung cancer who have failed regular chemotherapy9,10.Aberrantly activatedEGFRaffects various human cancers such as lung cancer, colorectal cancer, head and neck squamous carcinoma, and glioblastoma, among which glioblastoma has the highest rate ofEGFRgene alteration11-14.Over 50% of human GBMs show amplification,rearrangement, or point mutations inEGFR, and half ofEGFR-amplified tumors express the mutant receptor EGFRvIII, which causes constitutive activation of the receptor in a ligand-independent manner15. So far,EGFRvIII has not yet been detected in normal human tissue,thereby making it a potentially promising tumor-specific antigen16.
However, despite extensive preclinical and clinical efforts,EGFR inhibitors fail to produce significant clinical benefits in glioblastoma, and glioblastomas appear to be refractory to such inhibitors17-19. This intrinsic drug resistance is different from secondary drug resistance caused by long-term application of the same EGFR inhibitor in patients with nonsmall cell lung cancer. The underlying mechanism of the primary resistance of glioblastomas to EGFR inhibitors is largely unknown. Identifying the primary and intrinsic resistance mechanism of glioblastoma cells to EGFR inhibitors and finding ways to overcome it is of great value to recurrent glioblastoma patients with aberrantly activated EGFR signals who have no effective alternative treatment options. Therefore, this study used glioblastoma cell lines(U87MG and LN229) transfected with the EGFRvIII mutant(hereafter correspondingly referred to as U87vIII and LN229vIII cells), to explore the primary resistance mechanism of glioblastoma to gefitinib and identify ways to reverse this primary drug resistance.
Materials and methods
Cell lines and patient tissues
Human glioblastoma cell line U87MG and LN229 were obtained from American Type Culture Collection (ATCC).Lentivirus containing the EGFRvIII sequence and a negative control sequence was obtained from GenePharma and infected the two glioblastoma cell lines according to manufacturer's instructions. U87MG and LN229 cells stably expressing the EGFRvIII mutant were screened out with puromycin for two weeks and then used in the subsequent experiments. All glioblastoma cell lines were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% bovine calf serum (HyClone). Only early-passage cells were used in the study. No cell lines used in this study were found in the database of commonly misidentified cell lines that is maintained by ICLAC and NCBI BioSample. Fifteen cases of low-grade astrocytoma tissues and sixteen cases of glioblastoma tissues were from newly diagnosed glioma patients who were admitted to the Neurosurgery and Neurooncology Department of Tianjin Medical University Cancer Institute and Hospital from 2014 to 2016 with definite pathological diagnosis after surgery. All subjects signed informed consent forms, and this study was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital.
Reagents and primers
Gefitinib, temozolomide (TMZ), Axl inhibitor R428 and JNK inhibitor SP600125 were purchased from Selleck Chemicals,C87 from Tocris, and TNFα from Peprotech. Detailed antibody information used in this study was summarized in Supplementary Table S1. Human TNFα ELISA kit was obtained from Elabscience, and protein concentrating column was obtained from Millipore. Annexin V Apoptosis Detection Kit was from BD Biosciences. The quantitative real-time PCR (qRT-PCR) Kit was purchased from Takara.MTT was obtained from Sigma. qRT-PCR primers for GAPDH and TNFα were synthesized by Invitrogen Biotechnology, and the sequences have been previously reported20,21: GAPDH-F: 5′-GAAGGTGAAGGTCGGAGTC-3′; GAPDH-R: 5′-GAAGATGGTGATGGGATTTC-3′; TNFα-F: 5′-AGCCCATGTTGTAGCAAACC-3′; TNFα-R: 5′-TGAGGT ACAGGCCCTCTGAT-3′.
Cell survival
Cell viability was evaluated using MTT according to the manufacturers’ instructions. Glioblastoma cells (5-10 ×103/well) were seeded into 96-well plate and cultured in their corresponding medium for the indicated numbers of days.Cells treated with DMSO were used as controls. Cells were then incubated with MTT for 4 h. Viability was calculated as follows: Percentage viability = (ODexp- ODblankaverage)/(ODctlaverage- ODblankaverage)×100%.
Western blot
Glioblastoma cells were divided into control and several experimental groups. Different drug combinations were added and incubated for 24 or 48 h. Cells were then lysed in cold RIPA working buffer including protease/phosphatase inhibitor cocktail (Cell Signaling Technology) and PMSF.Frozen patients’ glioma tissues (approximately 50 mg per case) were resuspended in 500 μL cold RIPA working buffer and homogenized with a homogenizer. Thereafter, all protein samples followed the pretreatment protocol for western loading as we have previously reported22,23. 10 μg total protein/sample were separated using SDS-PAGE method.Then, all proteins were transferred to PVDF membranes and incubated with indicated antibodies. ECL substrate solution(Pierce) was dripped onto the membranes, and protein signals were captured on film (Kodak).
RNA extraction, reverse transcription, and quantitative PCR
Frozen patients’ glioma tissues (approximately 50 mg per case) were resuspended in 1 mL of TRIzol and homogenized using a homogenizer, and then followed by chloroform extraction, isopropanol precipitation, and ethanol washing.RNA was reverse transcribed into total cDNA according to the Reverse Transcription Kit instructions (Invitrogen). qRTPCR cycle parameters were as follows: 95°C for 10 s; 95°C for 5 s, and 60°C for 40 s, for a total of 40 cycles. HumanGAPDHwas used as the internal reference and ABI7500 built-in software was used for data analysis.
Animal studies
All mouse experiments were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University Cancer Institute and Hospital. Four- to six-weekold female athymic nude mice were purchased from Beijing Vital River Laboratory Animal Technology. Glioblastoma cells (1 × 106) were subcutaneously injected into the right flank of each nude mouse. When xenograft tumors were approximately 50 mm3, mice were randomly divided into control and experimental groups (6 mice for each group),and treated with the indicated drugs for 16 days. Tumor dimensions were measured using calipers every 2 days and tumor volumes were calculated with the formula: volume =(length × width2)/212. Mice were euthanized when tumor volumes exceeded 2000 mm3, or 16 days from the first day of treatment. Tumors from nude mice were fixed in 10%formalin and embedded in paraffin. Immunohistochemical staining was performed using the ABC streptavidin-biotin method with the SPlink Detection Kit (ZSGB-BIO) according to the manufacturer's protocol. Ki67 was scored as the percentage of nuclei-stained cells out of all cancer cells of hot spots in × 400 high-power fields; totally 500 to 1,000 tumor cells were counted in each case.
Statistical analysis
All data were analyzed for significance using GraphPad Prism 7.0. Each experiment was repeated at least three times. Unless otherwise indicated, all data were presented as mean ±SEMof three independent experiments. Two-tailed Student'sttest was used to compare two groups for independent samples.The results for statistical significance tests were included in the legend of each figure.P< 0.05 was considered statistically significant.
Results
Gefitinib treatment led to increased TNFα levels that triggered an adaptive pro-survival signaling pathway in glioblastoma cells

Figure 1 Gefitinib treatment led to increased TNFα levels that triggered an adaptive pro-survival signaling pathway in glioblastoma cells.(A) Immunoblotting of EGFRwt, EGFRvIII, and phospho-EGFR in glioblastoma cell lines stably transfected with EGFRvIII mutation. GAPDH was used as loading control. EGFRwt, EGFR wide-type. (B) U87vIII and LN229vIII cells were treated with indicated concentrations of gefitinib for 48 hours, and thereafter cell viability was tested by MTT assay. Data were mean ± SEM, n = 3 independent experiments. (C) U87vIII and LN229vIII cells were treated with 2 μM gefitinib for the indicated time intervals, and total EGFR, phospho-EGFR, total Axl, phospho-Axl, total JNK and phospho-JNK were detected by immunoblotting. (D) After U87vIII or LN229vIII cells were treated with 2 μM or 12 μM gefitinib for indicated time points, ELISA was introduced to detect the TNFα concentration in cell culture supernatant. U87vIII cells (E) or LN229vIII cells(F) were treated with 2 μM gefitinib for the indicated time intervals, and TNFα mRNA level was evaluated by qRT-PCR assay. In (D), (E) and(F), data were mean ± SEM, n = 3 independent experiments. Student’s t-test, *P < 0.05, **P < 0.01.
Although U87MG and LN229 cell lines are two of the most commonly used glioblastoma cell lines, their endogenous EGFR signal activation is very weak and both of them lack the EGFRvIII mutation. Thus, we overexpressed EGFRvIII in U87MG and LN229 to establish glioblastoma cell lines in which the EGFR downstream pathway was permanently activated in a ligand-independent manner, and used them asin vitrocell models to mimic the cytological behavior of glioma patients with the same mutation. Such cell models have already been widely used in glioma research field24-26. As shown in Figure 1A, EGFR mutation was successfully imported into U87MG and LN229 cells and the EGFR kinase activity was also significantly increased. Next, we evaluated the sensitivity of glioblastoma cell lines with EGFRvIII mutation to gefitinib treatment. As shown in Figure 1B,gefitinib lower than 12 μM had no obvious effect on the survival of LN229vIII or U87vIII cells. We added 2 μM gefitinib to U87vIII and LN229vIII cells for different time intervals and found that with time, phosphorylation of EGFR decreased, but the activation of the pro-survival intracellular kinase Axl and its upstream kinase JNK gradually increased(Figure 1C and Supplementary Figure S1A). This indicated that new pro-survival signals appeared to replace the weakened EGFR signal and thus, cells were able to survive.Actually, we also detected the activation of other survival closely related kinases such as Akt, STAT3 and p38 MAPK after gefitinib treatment, but their activity didn’t increase(Supplementary Figure S1B).
Considering that TNFα is known to be the strongest upstream activation factor of JNK kinase27,28, we used ELISA to assess the effect of gefitinib treatment on the expression of TNFα. As expected, gefitinib treatment induced a time- and dose-dependent increase in the concentration of TNFα in the cell culture supernatant of LN229vIII and U87vIII cells(Figure 1D). Moreover, the TNFα mRNA levels were upregulated after gefitinib treatment in U87vIII (Figure 1E)and LN229vIII cells (Figure 1F), indicating that the TNFα increase in cell culture may be due to transcriptional activation to some extent.
Together, these results suggest that primary resistance to gefitinib may be mediated by an adaptive TNFα-JNK-Axl signaling in glioblastoma.
High levels of TNFα in the glioblastoma microenvironment contributed to the primary resistance to EGFR inhibition
To determine if TNFα was sufficient to confer primary resistance to gefitinib in glioblastoma cells, we designed a reverse compensation experiment to detect whether the activation of TNFα signaling by addition of exogenous excessive TNFα could result in protection from cell death in gefitinib-treated glioblastoma cells. In this experiment, we used a gefitinib concentration of 25 μM, which induced statistically significant cell death in U87vIII and LN229vIII cells, and found that addition of exogenous TNFα protected these two cell lines from cell death induced by gefitinib(Figure 2A and 2B).
We collected 15 low-grade astrocytoma (WHO II-III) and 16 glioblastoma tissues, extracted mRNA from each, and compared theTNFαmRNA expression in the two patient groups using qRT-PCR. All patients were newly diagnosed glioma patients admitted to our department, who had no neo-adjuvant radiotherapy, chemotherapy, biotherapy, or other targeted treatment before surgery. The sex ratio and age span of the two groups were similar (Supplementary Table S2).As shown in Figure 2C, the expression ofTNFαwas significantly higher in the glioblastoma group than in the astrocytoma group (P< 0.001). Additionally, TNFα protein levels were consistent with the gene expression data, as TNFα protein was generally higher in the glioblastoma group than in the astrocytoma group (Figure 2D). In clinical practice,glioblastoma tissues are often accompanied by large areas of ischemic necrosis and inflammatory cell infiltration29,30, and TNFα is the most important and frequent regulator of the innate immune and inflammatory responses in the body31.Therefore, this may be one of the reasons for the high levels of TNFα in glioblastoma tissues. Together with previous studies, we infer that high TNFα levels in glioblastoma tissues may intensify the primary resistance of glioblastoma cells to gefitinib.
Therefore, the combination of a specific TNFα inhibitor and gefitinib may be a viable therapeutic strategy for overcoming primary EGFR-inhibitor resistance in glioblastoma, and provide improved clinical outcomes for glioblastoma patients.
TNFα inhibition with C87 sensitized glioblastoma cells to EGFR inhibition
A novel TNFα inhibitor, C87, as we have previously reported32, was screened out of a compound library of approximately 90,000 small-molecule compounds. C87 was identified as a hit compound using a combination of computer-aided drug design andin vitrocell-free and cellbased assays. C87 has been shown to effectively and specifically inhibit the biological activity of TNFα bothin vitroandin vivo, thus making it an ideal candidate for synergistic combination with EGFR inhibitors. Next, we explored whether the addition of C87 could increase the sensitivity of glioblastoma cells to gefitinib. Glioblastoma cells treated with a combination of a TNFα neutralizing antibody and gefitinib was used as a positive control. MTT results showed that C87, TNFα neutralizing antibody, or gefitinib (2 μM) alone did not induce significant cell death in U87vIII and LN229vIII cells, as cell viability was maintained above 85% (Figure 3A and 3B). However, gefitinib combined with C87 significantly decreased the viability of U87vIII cells to 45.6 ± 3.4% (P< 0.01,vs.gefitinib), and decreased the viability of LN229vIII cells to 41.3 ± 2.5% (P< 0.001,vs.gefitinib). The combination of gefitinib and the TNFαneutralizing antibody achieved similar results in U87vIII cells(P< 0.01,vs.gefitinib) and in LN229vIII (P< 0.01,vs.gefitinib). We also detected the apoptosis of U87vIII cells after gefitinib treatment with flow cytometry and the results further verified the combined effects of TNFα inhibitors and gefitinib (Figure 3C). From a more intuitive point of view,the morphological effect of TNFα inhibition in combination with gefitinib on glioblastoma cells was easily observed using light microscopy (Figure 3D). In that, control cells treated with vehicle, C87, TNFα-neutralizing antibody, or gefitinib alone maintained their morphology with long spindle-like shapes and clear edges. In contrast, more than half of the cells treated with a combination of C87 and gefitinib or gefitinib and a TNFα-neutralizing antibody underwent apoptosis, and cell debris was clearly visible in the microscopic fields.Additionally, gefitinib-induced activation of Axl kinase and JNK kinase was inhibited when U87vIII cells were treated with a combination of gefitinib and C87 or a combination of gefitinib and a TNFα-neutralizing antibody for 48 hours(Figure 3E). Together, these results showed that C87 combined with gefitinib could significantly increase the sensitivity of glioblastoma cells to gefitinib treatmentin vitro.

Figure 2 High levels of TNFα in the glioblastoma microenvironment contributed to the primary resistance to EGFR inhibition. After TNFα (1 ng/mL) alone, or gefitinib (25 μM) alone, or combination of the two treated U87vIII (A) or LN229vIII (B) cells for 48 hours, cell viability was tested by MTT assay. (C) The relative expression of TNFα mRNA in 15 low grade astrocytoma (WHO II-III) tissues and 16 glioblastoma tissues were detected with qRT-PCR method. In (A), (B), and (C), data were mean ± SEM, n = 3 independent experiments, Student’s t-test,*P < 0.05, **P < 0.01, ***P < 0.001. (D) Top: Immunoblotting analysis of protein levels of TNFα in the 15 low grade astrocytoma tissues (WHO II-III, serial number 1#-15#) and 16 glioblastoma tissues (serial number 16#-31#). Bottom: Western blot band intensity of TNFα was quantified and normalized to the internal control (mean ± SD, n = 3 independent experiments).
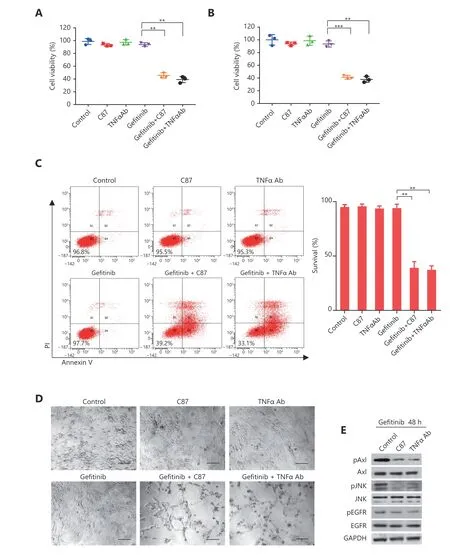
Figure 3 TNFα inhibition with C87 sensitized glioblastoma cells to EGFR inhibition. After C87 (2.5 μM), or TNFα antibody (1 μg/mL), or gefitinib (2 μM) alone, or combination treated U87vIII (A) or LN229vIII (B) cells for 48 hours, cell viability was tested by MTT assay. (C)Apoptotic assay of U87vIII cells with the above-mentioned treatments. In (A), (B) and (C), data were mean ± SEM, n = 3 independent experiments, Student’s t-test, *P < 0.05, **P < 0.01. (D) Representative morphology of U87vIII cells with the above-mentioned treatments,scale bar, 100 μm. (E) After U87vIII cells were treated with gefitinib combined with C87 or TNFα antibody for 48 h, expression of total Axl,phospho-Axl, total JNK, phospho-JNK, total EGFR and phospho-EGFR were detected by immunoblotting.
In order to prove the synergistic effect of C87 on gefitinib was indeed mediated by a concurrent inhibition of TNFα signaling, not by a simple combination of compound toxicity, we evaluated whether C87 could increase the sensitivity of glioblastoma cells to TMZ. As expected, TMZ treatment didn’t increase the TNFα mRNA expression in U87vIII or LN229vIII cells (Supplementary Figure S2A).Correspondingly, C87 failed to work synergistically with TMZ in the cytotoxic assay of glioblastoma cells(Supplementary Figure S2B). These results indicated from another perspective that the adaptive pro-survival TNFα signaling to some extent was unique and specific in the resistance of glioblastoma cells to gefitinib, and dual inhibition of EGFR and TNFα signaling might be a viable strategy to overcome gefitinib resistance.
We also explored the combined effects of gefitinib with Axl inhibitor or JNK inhibitor in U87vIII and LN229vIII cells. As shown in Supplementary Figure S3A and S3B, a concurrent blockade of Axl or JNK could sensitize these two cells to gefitinib treatment. Together with Figure 1C and Figure 3E,these results indicate that the TNFα-JNK-Axl signaling axis indeed plays an important role in the gefitinib resistance and inhibiting this axis at multiple nodes renders EGFRvIIImutant glioblastoma cells sensitive to EGFR inhibitors.
C87 sensitized mouse xenograft tumors to gefitinib monotherapy

Figure 4 C87 sensitized mouse xenograft tumors to gefitinib treatment. (A) U87vIII subcutaneous xenograft tumors were once a day treated with C87 (10 mg/kg) intraperitoneally or gefitinib (50 mg/kg) by oral gavage, or combination of the two. (B) LN229vIII subcutaneous xenograft tumors were treated daily with the same doses as in (A). In (A) and (B), tumor volumes were calculated as indicated in the Method. Values are mean ± SD, n = 6 mice for each group, Student’s t-test, ***P < 0.001 g (gefitinib group vs. gefitinib + C87 group). Inserts were representative tumor photos of gefitinib group or gefitinib + C87 group. (C) U87vIII (top) or LN229vIII (bottom) subcutaneous xenograft tumors of each group were resected, fixed in formalin, and stained for Ki67. Scale bar, 100 μm.
Thein vivotherapeutic potential of the combination of gefitinib and the TNFα inhibitor C87 was examined using subcutaneous glioblastoma xenograft models. The experiment was conducted by injecting U87vIII or LN229vIII cells into the right flank of athymic mice. Once subcutaneous tumors became visible, the mice were divided into four treatment groups: control, gefitinib alone, C87 alone, or gefitinib+C87. While gefitinib and C87 as monotherapy modestly reduced tumor growth, the combination was significantly more effective than either compound alone(Figure 4A and 4B). Additionally, the combination was more potent than the single agents at inhibiting tumor cell growth,as shown by immunohistochemical staining of Ki67(Figure 4C and Supplementary Table S3). For example, the combination group significantly decreased the Ki67 index of U87vIII tumors to 9.0±4.6% (P< 0.01,vs.gefitinib), and decreased the Ki67 index of LN229vIII tumors to 12.6±3.9%(P< 0.01,vs.gefitinib). Collectively, ourin vivoobservations recapitulated thein vitroresults, suggesting that concurrent inhibition of TNFα has the potential to significantly improve the efficacy of EGFR inhibitors for treating EGFR-positive glioblastoma patients.
Discussion
Epidermal growth factor receptor (EGFR), the coding product of the proto-oncogeneC-ErbB-1, is a transmembrane protein33,34. The EGFR signaling pathway plays an important role in the proliferation, survival,invasion, metastasis, and angiogenesis of tumor cells35,36.EGFR signaling is highly activated in many epithelial cellderived tumors, such as non-small cell lung cancer, glioma,head and neck cancer, and colorectal cancer12,37. EGFR inhibitors (EGFR-TKIs) are highly effective in the treatment of EGFR-positive non-small cell lung cancer and other tumors10,38. However, with extended use, secondary drug resistance to EGFR-TKIs is acquired, and the disease progresses rapidly after drug resistance39. Currently, there are two schools of thought for this kind of secondary drug resistance: one is the emergence of the EGFR-T790M mutation in tumor cells after long-term treatment with EGFR-TKIs, which activates Axl, Met, or other kinases,leading to the restoration of pro-growth signals40; the other one is that, after long-term inhibition of EGFR signaling,tumor cells increase the activity of the STAT3 signaling pathway to resist the death signals elicited by EGFR inhibition41. However, little is known about why most EGFRpositive glioblastoma patients do not appear to be oncogene addicted, and EGFR TKIs, so far, have not been effective in this patient population, even in newly diagnosed patients.
Gefitinib is the most commonly used EGFR-TKI in China,and is the preferred EGFR-TKI of choice because of its efficacy and price. In our study, we explored the mechanism of primary drug resistance to gefitinib in glioblastoma cells.We found that the primary resistance of glioblastoma cells to gefitinib may be related to the rapid activation of intracellular TNFα signaling after drug administration, and with clinical samples, we also confirmed that the high expression of TNFα in glioblastoma tissues further intensified gefitinib resistance.Additionally, C87, as a novel inhibitor which could competitively and specifically interrupt the binding between TNFα and its receptor and thus block the TNFα downstream signaling, sensitized glioblastoma cells to gefitinib treatment bothin vitroandin vivo(summarized in Figure 5).Therefore, dual blockade of TNFα and EGFR may provide a new therapeutic strategy for patients with advanced EGFRpositive glioblastoma who have failed conventional chemotherapy regimens.
Existing TNFα inhibitors (e.g., infliximab, adalimumab,cimzia, and enbrel) are all water-soluble macromolecular protein preparations of antibodies or receptors and have already achieved great success in the treatment of some autoimmune diseases (rheumatoid arthritis, inflammatory bowel disease, etc.)42-44. However, their physical structures result the poor stability and the exclusion from blood-brain barrier. Small-molecule TNFα inhibitors such as thalidomide also have strong off-target pharmacological activity and thus,serious side effects45. Guo et al.24have demonstrated that the primary resistance of established glioblastoma cell lines to erlotinib could be overcome by combining Sp600125 (JNK inhibitor) or thalidomide with erlotinib. In our study, we treated glioblastoma cells with a combination of the TNFα specific small-molecule inhibitor C87 and gefitinib. We also used a combination of gefitinib and a TNFα-neutralizing antibody as a positive control. The results showed that C87 could significantly increase the sensitivity of glioblastoma to gefitinib bothin vitroandin vivo, and thein vitroeffect was comparable to that of the TNFα-neutralizing antibody. With deference to the blood-brain barrier, the combination of C87 and gefitinib for the treatment of glioblastoma may have greater clinical advantages and more potential for clinical translation than existing TNFα inhibitors.
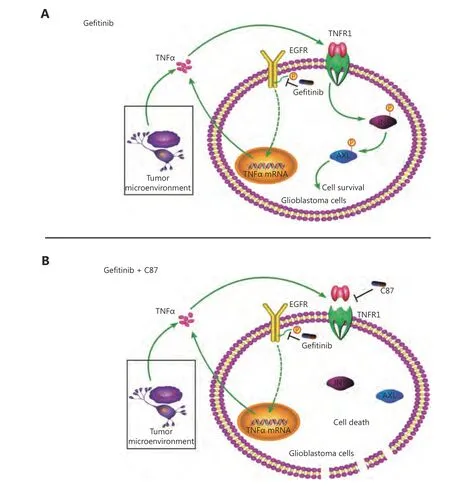
Figure 5 A diagrammatic sketch illustrates how C87 overcomes the resistance of glioblastoma cells to gefitinib. (A) In glioblastoma cells treated with gefitinib alone, the pro-survival TNFα signaling pathway, either derived from the irritated tumor cells themselves, or tumor environment, enables the cells to bypass cell death induced by EGFR inhibition. (B) C87, as a novel inhibitor which competitively and specifically interrupts the binding between TNFα and its receptor and thus blocks the TNFα downstream signaling, induces glioblastoma cell death together with gefitinib.
Although the roles and signaling pathways of TNFα in innate immune and inflammatory responses have been extensively defined46,47, the relationship between TNFα and downstream signaling mechanisms in tumor cells has not been well established. Numerous studies have shown that TNFα in the tumor microenvironment not only fails to kill tumor cells, but also plays an important role in the occurrence and development of tumors48,49. For example,TNFα can promote the proliferation, malignant transformation, angiogenesis, invasion, and metastasis of tumor cells50,51. The roles of cytokines, including TNFα, in glioblastoma had long been noticed52. However, the roles of TNFα in the tumorigenesis and development of glioma failed to attract considerable attention, partially due to the“immune privilege” characteristic of brain53and the complexity of TNFα function54. Kusne et al.25once reported that the myeloid cell-derived TNFα promotes glioblastoma cell proliferation, motility and resistance to erlotinib through a RTKs-aPKC-NFKB pathway. Our study also supports that TNFα in glioblastomas may induce primary drug resistance to gefitinib, and the TNFα specific small-molecule inhibitor C87 is able to significantly increase the sensitivity of glioblastoma to gefitinib. Interestingly, tumor growth was slightly slower in tumor-bearing mice treated with C87 alone in comparison to cohorts treated with vehicle. This indicated that TNFα signaling itself might play a role in glioblastoma development. In summary, this study has provided vital information about the relationship between inflammatory factors represented by TNFα and brain tumorigenesis.
Conclusions
Overall, our study showed that the primary resistance of glioblastoma cells to gefitinib may be related to the rapid activation of intracellular TNFα signaling after drug administration, and with clinical samples, we also confirmed that the high expression of TNFα in glioblastoma tissues further intensified gefitinib resistance. Furthermore, C87, as a novel inhibitor which could competitively and specifically interrupt the binding between TNFα and its receptor and thus block the TNFα downstream signaling, sensitized glioblastoma cells to gefitinib treatment bothin vitroandin vivo. Therefore, dual blockade of TNFα and EGFR may provide a new therapeutic strategy for patients with advanced EGFR-positive glioblastoma who have failed conventional chemotherapy regimens.
Acknowledgments
We thank Department of Public Laboratory of Tianjin Medical University Cancer Institute and Hospital for their help in equipment and technical support. This work was supported by National Basic Research Program of China(Grant No. 2015CB964903), Natural Science Foundation of Tianjin (Grant No. 15JCQNJC44800 and 18JCQNJC81300),National Natural Science Foundation of China (Grant No.81702481, 81701224, 81802873 and 81600083), CAMS Innovation Fund for Medical Sciences (Grant No. 2016-12M-1-003; 2017-12M-1-015) and the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Grant No. 2017PT31033, 2018RC31002,2018PT32034).
Conflict of interest statement
No potential conflicts of interest are disclosed.
 Cancer Biology & Medicine2019年3期
Cancer Biology & Medicine2019年3期
- Cancer Biology & Medicine的其它文章
- A four-gene signature-derived risk score for glioblastoma:prospects for prognostic and response predictive analyses
- Prediction of cervical lymph node metastases in papillary thyroid microcarcinoma by sonographic features of the primary site
- Decrease in the Ki67 index during neoadjuvant chemotherapy predicts favorable relapse-free survival in patients with locally advanced breast cancer
- Incidence, distribution of histological subtypes and primary sites of soft tissue sarcoma in China
- Prevalence and clinical significance of pathogenic germline BRCA1/2 mutations in Chinese non-small cell lung cancer patients
- Incomplete radiofrequency ablation provokes colorectal cancer liver metastases through heat shock response by PKCα/Fra-1 pathway