
GMP计算机化系统审计追踪审核探讨
2019-09-24 18:30:46路佳特
上海医药 2019年15期
路佳特
摘 要 数据可靠性是近年来世界各国药品监管机构的检查重点,也是发现缺陷项比较多的领域。数据可靠性中比较关键的功能或检查点是审计追踪,本文从欧美法律法规出发,对计算机化系统和关键数据进行分类,基于风险评估和统计学角度,明确审计追踪审核的方法。审计追踪记录的信息很多,应该把有限的资源花在重要和风险高的信息上,保证审核的可操作性和科学性。
关键词 药品生产质量管理规范 计算机化系统 数据可靠性 审计追踪
中图分类号:R951 文献标志码:C 文章编号:1006-1533(2019)15-0068-04
Discussion of auditing trail of GMP computerized system
LU Jiate
[Merck Pharmaceutical Manufacturing (Jiangsu) Co., Ltd., Jiangsu Nantong 226010, China]
ABSTRACT Data integrity has been the focus of drug regulatory agencies around the world in recent years, and is also the field where there are more defects. The key function or check point in data reliability is audit trail. The computerized systems and key data were classified based on European and American laws and regulations and meanwhile the method of audit trail review was defined based on risk assessment and statistics. Audit tracking records have a lot of information, we should spend limited resource on important and high-risk information so as to ensure the operability and scientific nature of audit trail review.
KEy WORDS GMP; computerized systems; data integrity; audit trail
数据可靠性越来越多地被各个国家药品管理机构提及和检查,审计追踪是检查过程中的重点,审计追踪的执行情况也直接反应药厂数据可靠性的管理水平。审计追踪只是系统或是人为的记录,作为数据的拥有人,药厂平时应该使用科学合理的方法审核审计追踪,以便符合法规要求,并提前发现问题。笔者从各个国家的法规和指南出发,结合工作实际经验,探讨比较合理和可执行的审计追踪审核方法。
1 国內外法规指南
欧盟GMP附录11中提及,要基于风险评估考虑使用审计追踪功能,创建一份可以包含GMP相关信息的变化和删除的记录。变化和删除的原因也应当被记录下来。审计追踪应当适用于且可转化为通常可理解的样式和定期性的审核。
美国FDA 21 CFR Part 11中提及,对于封闭系统的控制,也就是具备权限控制的系统。那些在其中创建,修改,维护或传输电子记录的人员应采用旨在确保电子记录的真实性,可靠性和适当的机密性的程序和控制,并确保签名者的签名与签名人是直接关联的。所有人在封闭系统中所做的事,都应该可以准确追溯到和记录到谁,什么时间,做了什么事,原因是什么。
此类程序和控制措施应包括使用安全的,计算机生成的带时间戳的审计追踪来独立记录操作员条目的日期和时间以及创建,修改或删除电子记录的操作。记录更改不得模糊以前记录的信息。此类审计追踪文件的保留期限应至少与主题电子记录所要求的时间一致,并应可供机构审查和复制。
中国《药品数据管理规范(征求意见稿)》中提及,使用计算机化系统创建、更改数据等操作,应当通过审计追踪功能记录,确保其可追溯性。现有计算机化系统不具备审计追踪功能的,可以使用替代方法,如日志、变更控制、记录版本控制或原始电子记录辅以纸质记录来满足数据可追溯性的要求。不得关闭计算机化系统的审计追踪功能,不得修改审计追踪产生的数据。应当对审计追踪进行审核,审核的频率和内容应当基于风险级别确定。涉及直接影响患者安全或产品质量的关键数据更改(如最终产品检验结果、测试样品运行序列、测试样品标识、关键工艺参数的更改等),至少应当在做出最终决定前对更改的数据及其审计追踪一并进行审核。
国际制药工程协会(ISPE)的良好自动化生产实践指南的记录和数据可靠性指导原则(GAMP records and data integrity guide)中建议,审计追踪审查应建立并记录,应作为例行数据审查/批准过程的一部分,应由生成数据的操作区域执行。审计追踪审查的要求(例如频率,严谨,角色和职责)应基于书面化的风险评估,考虑到业务流程,数据的关键性,系统的复杂性及其预期用途,对产品质量和患者安全的潜在影响。
由此可见,国内外的法规和指南文件中都提到过审计追踪,都较为明确地指出要审核审计追踪及审核的内容,并且要基于风险评估审核范围和审核频率。
2 方法
2.1 审计追踪审核流程
审计追踪审核流程可以分为:定义数据的关键程度,确认计算机化系统审计追踪功能,风险评估,分级审核审计追踪,回顾和自检(图1)。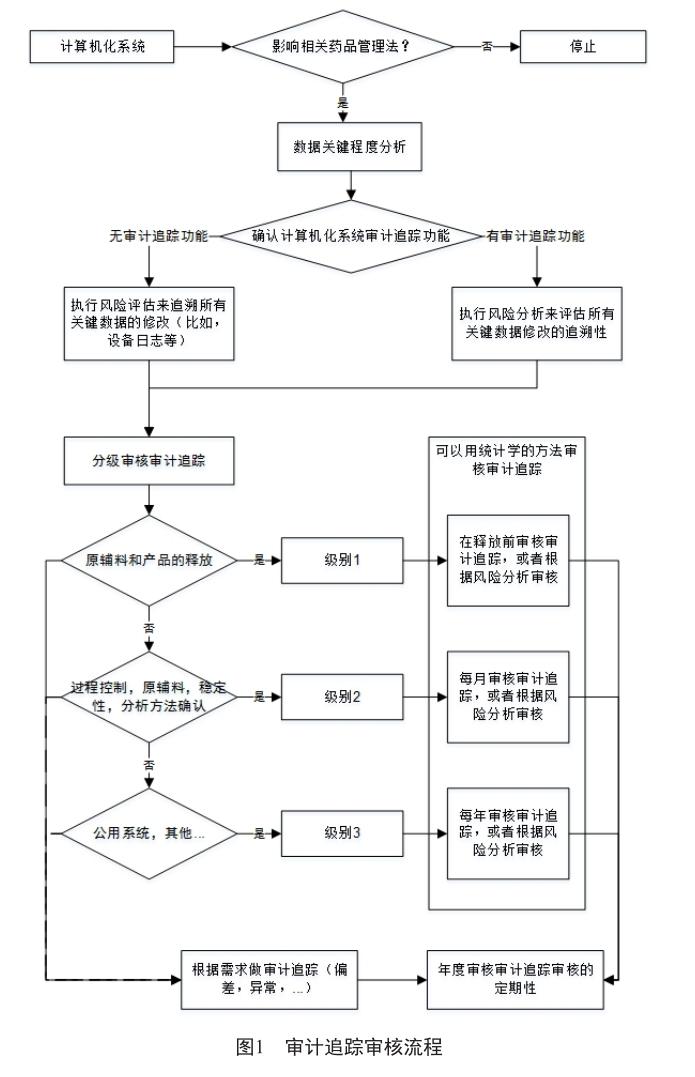
2.2 定义数据的关键程度
GMP计算机化系统可以记录的信息有很多,审计追踪中包含的内容也是包罗万象,小到人员登入登出,大到关键工艺参数的修改。我们首先应该定义哪些数据是关键数据。欧洲药品管理局(EMA)在一份数据可靠性的问答中提到如何评估数据的关键性,受数据影响的决定可能在重要性上不同,并且数据对决策的影响也可能不同。有关数据关键性的考虑因素包括:第一,数据用于做什么样的GMP决策?例如:最终产品检测结果,比产品中间过程检测的结果要重要,确定符合关键质量属性的数据比仓库清理记录更重要。第二,数据对产品质量或安全性有何影響?例如:对于口服片剂,活性物质测定数据对产品质量和安全性的影响比片剂尺寸数据更大。我们可以基于以上两个原则,对系统中每一种GMP数据进行关键程度分级和比较,来定义哪些数据可以作为相对关键数据审核(表1)。
2.3 确认计算机化系统审计追踪功能和风险评估
对于计算机化系统进行筛查,确定其是否有审计追踪功能。如果没有审计追踪功能,需要执行风险评估,降低所有关键数据没有被记录的风险,找出合适的方法记录对关键数据的创建,修改和删除。
如果系统具备审计追踪功能,也需要进行风险评估,识别系统记录审计追踪的风险,定义合适的行动确保所有关键数据的审计追踪能够及时、方便、全面审核。比如,所使用的校准曲线的号码没有提供审计追踪,并且这些信息被认定为关键性的,则应该采取一些缓解措施。风险评估的方法可以选择FMEA的方式,邀请各个领域的专家一同评估。如有必要,可以邀请供应商一起参与评估。评估的结果都要经过QA的批准,并且落实到系统或者设备的SOP中。
2.4 分级审核审计追踪
找出所有关键数据,并且确定合适的审计追踪记录方式,对审计追踪的审核进行分级管理。因为关键程度不同,风险就相应不同,所以可以配置不同的资源在审计追踪的审核上。分级的要求大致与数据关键程度分级类似,可以定义3个级别,分别对应3个审核频率(表2)。
表2只是粗略地给出了一个大致的频率分类,也可以按照统计学的方法制定审核策略,同样具备科学性和代表性。数据应由数据创建部门审查,必要时经QA复核。
2.5 回顾和自检
按照既定的程序执行审计追踪的审核,审计追踪审核期间发现的任何重大偏差必须全面调查和记录。如果对审计追踪的审核发现可能影响产品质量的问题/差异,就必须制定程序来定义要采取的行动。反之,如果日常GMP活动发现了偏差,也可以反过来到系统中查看审计追踪的记录,从而帮助找出偏差的根本原因。如果日常发生的变更导致数据关键程度,系统配置和流程的因素发生了改变,那么涉及到的审计追踪也应该评估其适用性,如果需要,检查的条目或频率可以改变。
QA应该对审计追踪审核的情况进行监督或者复核,以确保审计追踪审核是按照已批准的流程执行的。同时,QA必须制定一个计划和时间表,根据其重要性对审计追踪进行持续性审查。该计划必须成为自检计划的一部分,自检活动必须形成文件并记录。
3 讨论
分级审核审计追踪可以合理地关注重点数据,科学地分配资源,以达到在合规的情况下,尽可能利用合适的资源,确保执行的有效性。
审计追踪审核是一项系统工程,需要具备专业知识的人一起协作,找出一个既可以降低风险,又能满足法规要求的科学合理的方法。
