
多核金属配合物合成及其催化性能研究进展
2019-09-24 09:12刘婷婷陈辉杨振强杨瑞娜
应用化工 2019年9期
刘婷婷,陈辉,杨振强,杨瑞娜
(河南省科学院化学研究所有限公司,河南 郑州 450002)
近年来,随着对配位化学研究的深入,多核金属配合物在均相催化与材料化学等研究领域受到越来越多的关注[1],具有特殊物理、化学及生物等性能的多核金属配合物取得了快速的发展[2]。多核金属中心之间体现出的协同效应,能使其展现出更加优越的催化性能[3]。科学家们对金属之间的协同作用的研究主要是受到自然界中酶的作用模式的启发。在自然界生物体系中,通过彼此接近的多个反应中心协同活化和催化是酶催化采取的普遍策略[4-8]。两个反应物通过配位或者非共价键作用处于最优反应位点是酶催化具有出色的效率和无与伦比的选择性的关键因素。在多中心金属配合物催化剂的催化过程中,多个金属中心同时作用于底物,通过多金属协同效应来实现化学键的断裂与形成。
1 双核金属配合物
1.1 同核双金属配合物
1999年,Sita等[9]首次报道了二亚胺插入金属碳键的反应,合成了基于脒基骨架的双核钛配合物,产物结构通过X-射线单晶晶体衍射技术得到确定。变温核磁实验发现这种基于脒基骨架的双钛配合物在溶液状态存在rac-和meso-两种构象。2008年,他们合成了一系列不同烷基链的脒基双锆配合物[10]。研究发现双核锆配合物在硼酸盐作为助催化剂的条件下,可以高效、高化学选择性的实现丙烯聚合反应。研究结果表明,随着两个金属原子之间距离的减小,反应的选择性逐渐降低。原因可能是随着两个金属原子之间距离的降低,使得催化活性中心周围的位阻越大,导致反应的选择性下降。
Marks课题组[11]利用桥联双茚基配体合成了一系列单、双金属锆和钛金属配合物,并系统性地研究了双金属配合物中两个金属之间的协同效应对催化乙烯均聚反应以及乙烯和苯乙烯共聚反应的影响,结果显示双金属配合物比相应的单金属配合物具有更高的催化活性。同时,他们也提出了双金属协同催化作用的机理过程,双核金属中心与共聚中间体协同作用,实现共聚链的快速增长,是其催化活性优于单核金属配合物的重要原因。

近年来,基于手性ProPhenol配体的双核锌配合物[15]被广泛应用于不对称加成反应中,具有较广的底物适用范围。潜亲核试剂如恶唑酮、2-呋喃酮、硝基烷烃、吡咯类、炔烃化合物等在手性锌配合物催化剂的作用下与亲电试剂如醛基化合物、亚胺、硝基烯烃、酰基硅烷类化合物、苯甲酸乙烯酯、α,β-不饱和羰基化合物等反应得到一系列对映体过量的化合物,且基于ProPhenol配体的催化剂可用于合成20种以上的天然产物。利用这种配体替换传统的手性助剂、烯醇或者硅烷试剂等应用于催化aldol、Mannich及Henry反应,具有更高的原子经济性。因此,ProPhenol体系是对有机催化的进一步完善。
Yu研究组利用组装及基于配体合成策略发展了两种双核钌金属配合物[16-17],并应用于催化酮转移氢化反应。其中基于组装合成策略得到的双核钌金属配合物,获得了最高1.4×107h-1的半程TOF值,是目前报道的酮转移氢化反应活性最高的体系之一。他们认为双核钌之间的协同催化作用是此催化体系展现优越催化活性的重要原因之一。
1.2 杂核双金属配合物
相对于同核双金属配合物,异核双金属配合物的研究相对较少。Crimmin研究组首次报道了通过氢原子相互连接的异核Au-H-Cu金属配合物[18]。在异核双金属催化剂的作用下,CO2与HBpin反应可以选择性生成HCO2Bpin。实验证明,金与铜对于催化该反应都是必须的,单一的金属催化剂催化该反应时,反应性与选择性均较差,从而证明了协同催化在有机催化反应中的应用及其重要性。
Shibasaki小组[19-20]合成了Shiff碱型异核双金属配合物,这两种双核金属配合物的不同点在于配合物采用Cu和Sm作为金属中心,而另一配合物采用Pd和La作为配位金属。配合物Cu/Sm催化亚胺的硝基-曼尼希反应主要得到顺式加成产物,只在铜催化剂作用下不能催化该反应的进行,而相应的Sm催化剂只有14%的转化率,远低于异核双金属配合物催化剂Cu/Sm的催化结果。配合物Pd/La催化醛类化合物的Nitroaldol反应则主要得到反式加成的产物,相比于异核双金属催化剂Cu/Sm,催化剂Pd/La则能表现出更高的催化反应活性及选择性。
2010年Peris小组[21],他们合成了异核双金属Ir/Pd化合物,以其作为催化剂,研究了芳香族硝基化合物与伯醇类衍生物之间的缩合反应。实验结果表明,异核Ir/Pd卡宾化合物可以较好地催化芳香族硝基化合物与芳香族伯醇类衍生物形成亚胺的反应。单独铱或钯的金属前体在催化该反应时,转化率较低;而将两种金属混合应用于此催化反应,也能得到较好的反应结果,但相比于异核Ir/Pd卡宾配合物催化剂,不仅底物适用性较低,而且需要较高的催化剂用量。这可能归因于金属铱与活性钯物种在硝基的还原过程中相互作用,开辟了新的反应路径。另外,以异核Ir/Pd卡宾配合物作为催化剂,在类似的反应条件下,能以77%的产率实现苄醇与对溴硝基苯的氧化偶联和Suzuki-Miyaura偶联的一锅多步串联反应,为合成联苯类亚胺衍生物提供了新的合成方法。
2 三核金属配合物
2.1 同核三金属配合物
2016年,孙立成研究组制备了双核钌配合物及三核钌配合物[22],在水氧化反应中,三核的钌配合物相比于双核钌配合物及单核钌配合物表现出更高的催化活性,能得到更高的TON值及更高的氧气初始释放速率。其中三核钌配合物催化剂催化水氧化的TON值为双核钌配合物TON值的2倍以上。动力学研究表明,双核与三核金属配合物在O—O键的形成过程中为分子内的作用,所以增加催化剂的活性位点有利于在分子内形成O—O键,从而提高催化效率。
Ema研究组通过Stille偶联反应成功合成了双官能团化的多卟啉催化体系[23],与相应的单核催化体系相比,多卟啉金属配合物催化剂在催化二氧化碳对环氧化合物的插入反应合成碳酸酯的反应中表现出更好的催化活性。当金属中心为镁时,能得到220 000的TON值和46 000 h-1TOF值;当金属中心为锌时,得到310 000的TON值和40 000 h-1的TOF值,是当时报道的由二氧化碳和环氧化合物合成环状碳酸酯的最高催化体系之一。他们认为反应位点之间的相互作用及对底物的同时活化是多核金属配合物催化剂表现高活性的重要原因。Nomura研究组报道了金属钛配合物催化的乙烯基聚合反应[24],多核钛配合物相较于单核钛配合物表现出更好的催化反应活性。
Bharadwaj报道的三核钴配合物催化剂也可以应用于催化二氧化碳对环氧化合物的插入反应[25]。该研究组由L-脯氨酸制备了氮杂-氧杂的穴状配体,此穴状配体可用作手性有机催化剂高活性催化醛与酮水相中的Aldol反应。穴状配体与Co前体配位后可形成三核的Co配合物,并获得了其单晶结构。Co配合物催化剂与共催化剂Bu4NBr相互作用可高效催化CO2与环氧化合物反应合成环状碳酸酯。在三核钴催化剂作用下,将环状碳酸酯的合成实现工业化,可极大程度地吸收空气中的二氧化碳,有利于减少温室效应。
2.2 杂核三金属配合物
Ishitani研究组通过Mizoroki-Heck反应再对连接配体进行光化学氢化合成了两种三核Os(II)-Re(I)-Ru(II)配合物催化剂[26]。此配合物催化剂在光作用下成功催化二氧化碳还原为一氧化碳,其中Os与Ru中心在反应中作为氧化还原光敏剂,Re中心则作为催化剂催化反应进行。3种金属相互作用相比于只有两种金属作用时,提高了光催化剂作用的持久性,使得催化效率由27%提高到55%,将TON值由854提高到4 347。
van der Vlugt研究组成功制备了三核的Au-Ni-Au金属配合物[27],并展现了优越的电化学性能,根据循环伏安法及控制电位库仑法研究发现,只有三核的金属配合物才能成功催化将苄基溴苯转化为1,2-二苯基乙烷,双核的Au-Ni配合物则不能催化此反应的进行。并研究发现三核金属金-镍之间的相互作用是能实现电催化反应构建C—C键的至关重要的因素,从而侧面证明了协同催化的重要性。
3 多核金属配合物
3.1 同核多金属配合物
多金属配合物催化剂由于多个彼此接近的中心原子之间的独特相互作用,能通过全新的模式来活化底物的反应位点,从而使反应表现出更高的反应速度及立体选择性,大量具有高活性的多核金属配合物催化剂被报道。

Hey-Hawkins研究组利用环状P4Mes4C(NCy)配体与[AuCl(tht)](tht=四氢噻吩)反应合成了十六环的Au-P超分子配合物[29],并通过核磁共振、高分辨质谱及X射线单晶衍射确定了结构,对于其它方面的研究目前还在进一步研究中。
3.2 杂核多金属配合物
Ruiz研究组[30]利用环戊二烯基钛单核前体与巯基吡啶锂盐作用合成巯基吡啶连接的环戊二烯基钛配合物,吡啶基配位点进一步与铂或者钯的配合物组装合成异核的钛铂或者钛钯配合物,并对其进行核磁表征及DFT计算确定了多核配合物的结构。分子动力学模拟表明,Ti—S键比M—Npy (M=Pd,Pt)键更倾向于进行配位-解离过程,这可能是前期过渡金属与硫原子软硬匹配的结果。其催化应用目前还在进一步研究中。
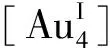
4 结论
本文综述了不同类型的多核金属配合物的合成及对其催化性能的研究。结果表明,多核金属配合物之间的协同作用是其展现出优于单核金属配合物催化活性的重要原因之一。虽然协同作用的模式还在进一步的探索中,但对多核金属配合物在催化、材料、生物等领域的应用研究将会越来越深入。
猜你喜欢
化工管理(2022年13期)2022-12-02
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
房地产导刊(2022年1期)2022-02-28
云南化工(2020年11期)2021-01-14
航天工业管理(2020年9期)2020-12-28
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
重型机械(2020年2期)2020-07-24
分析化学(2017年9期)2017-10-16
计算机测量与控制(2017年6期)2017-07-01
