
卷曲霉素抗结核作用及耐药机制的研究进展
2019-09-19 11:26官政燕
中国人兽共患病学报 2019年8期
官政燕,刘 梅,陈 玲
结核病(Tuberculosis,TB)是由结核分枝杆菌(Mycobacteriumtuberculosis, MTB)感染引起的慢性呼吸道传染病,是全球十大死亡原因之一。目前,耐多药结核病(multidrug-resistant tuberculosis,MDR-TB,即MTB至少对利福平和异烟肼同时耐药)及广泛耐药结核病(extensively drug-resistant, XDR-TB,即MTB不仅对利福平和异烟肼同时耐药,也对氟喹喏酮类药物中任何一种药物以及3种二线抗结核注射药物卷曲霉素、卡那霉素和阿米卡星中至少1种具有耐药性)的出现和传播,对全世界结核病的有效控制构成重大威胁。据世界卫生组织(World Health Organization, WHO)估计,2017年全球新发结核病1 000万例,MDR-TB占46万,其中约8.5%的MDR-TB患者为XDR-TB[1]。全国第5次结核病流行病学抽样调查报告结果显示,我国结核病总耐药率高达36.8%,耐多药率为6.8%[2]。适当使用二线注射药物(卡那霉素、阿米卡星或卷曲霉素)有助于治疗MDR-TB和预防XDR-TB[3]。卷曲霉素(Capreomycin, CPM)是一种环肽类抗生素,被认为是治疗MDR和MTB潜伏感染的关键二线注射类药物[3-4],其主要作用机制是干扰核糖体的功能,抑制蛋白质的合成[5-7]。然而,随着临床上越来越多的使用,CPM耐药问题逐步浮现,研究发现CPM耐药性主要与其作用靶点相关基因的突变有关[8-16]。因此,深入了解CPM的作用机制及耐药机制,有助于指导临床合理用药,延缓其耐药性的传播以及有效防治MDR-TB。
本文就CPM治疗结核病的作用机制、耐药机制及相关基因突变的研究进展进行综述,以期为CPM的临床应用提供参考依据,促进耐药基因检测技术的发展和加速抗结核新药物的开发进程。
1 抗菌作用机制
CPM于1979年应用于抗结核治疗,是治疗耐多药结核病的重要二线药物,对非复制MTB也有杀菌作用[4]。其主要抗菌作用机制是干扰核糖体功能,从而抑制蛋白质的合成;还可通过改变细菌细胞壁的结构,影响生物膜的发育;此外,还与金属离子协同作用发挥其抗菌活性。
1.1抑制蛋白质的合成 研究发现CPM通过tlya的甲基化作用结合到23S rRNA螺旋H69的核苷酸C1409区域和16S rRNA螺旋H44的核苷酸C1920区域之间形成的亚基间桥b2a上,影响细菌核糖体蛋白质的合成[11]。研究表明当表达不同的tlya同源物(tlyaI或II)时,大肠杆菌重组体对CPM的敏感度不同,其具体机制有待进一步研究[6]。近期研究显示,tlya能与s-腺苷-甲硫氨酸(S-Adenosyl-L-Methionine, SAM)结合,发挥其甲基化作用,加强MTB对CPM的敏感性[5]。MTB核糖体蛋白L12和L10是50S核糖体的组成部分,在翻译过程中L12-L10与伸长因子EF-G和EF-Tu的结合使GTP酶活性增强,使核糖体蛋白质合成增加[17]。研究表明CPM能干扰L12-L10的相互作用,破坏其与伸长因子EF-G和EF-Tu的结合,削弱GTP酶的活性,导致核糖体依赖的蛋白质合成受阻,还发现L10和(或)L12的过表达能降低CPM的抗菌活性,因此推测核糖体蛋白L12和L10很可能是CPM在体内作用的新靶点[7]。
1.2改变细菌细胞壁的结构、影响生物膜的发育 SirR是由Rv2788编码的一种转录抑制因子,研究发现Rv2788能下调CPM耐受相关基因(Rv0039,Rv0812,Rv1286)的转录,增强耻垢分枝杆菌对CPM的敏感性,而这些基因多与细胞壁相关。因此,推测SirR可能通过调控相关基因的转录水平,进而改变细胞壁结构,调控对CPM的敏感性[18]。此外,有研究发现噬菌体swu1 A321_gp67(一种假定的gtp酶激活蛋白)过表达使重组耻垢分枝杆菌对CPM等抗细菌药物极其敏感,进一步研究发现gp67过表达能引起细胞壁和生物膜发育相关基因MSMEG_0235,MSMEG_6092,MSMEG_1876和MSMEG_0382的表达下调,从而改变耻垢分枝杆菌的细胞壁结构,破坏生物膜的形成。因此,推测gtp酶激活区可能起一定作用,且提出gp67可以作为一种增效剂加入到现有的抗生素方案中,以更好地控制MTB的耐药菌株,然而,噬菌体组分如何抑制这些基因的转录目前还不清楚[19]。
1.3CPM与金属离子的协同作用 CPM富含氮的多肽结构使其能与其他金属离子特异结合,特别是铜离子,被认为是一种Cu2+的螯合剂[20],通过对铜-CPM复合体的研究发现,Cu2+能够影响分子的极性,将Cu2+附着在药剂上可以阻止其与多余的氢键及蛋白质中的无关阳离子的结合,在适宜的pH条件下,铜-CPM复合体组比纯CPM组对MTB的抗菌活性提高250倍。因此,认为可将Cu2+作为CPM等抗生素的载体[21]。
2 耐药机制
现有的研究普遍认为CPM的耐药机制主要是tlya、rrs基因发生突变导致药物作用靶点的改变。此外,eis的突变也可导致CPM耐药。已有报道CPM与氨基糖苷类药物之间存在交叉耐药性[12,22-24]。
2.1tlya基因 研究发现tlya通过调节细菌核糖体的甲基化,增强CPM的抗菌作用[6,11],而tlya基因的突变使核糖体缺少了甲基化,导致耐药性的产生[10-11]。通过体外诱导tlya基因的突变位点为野生型,可恢复耐药菌株对 CPM的敏感性[10],在大肠杆菌中重组tlya的表达明显增加对CPM的敏感性[11],验证了tlya基因突变导致CPM耐药的发生。tlya突变主要导致CPM低水平耐药[13],可引起CPM和紫霉素交叉耐药[22],突变位点见表1。但以往报道的tlya突变主要存在于体外筛选的耐药突变株中[8,10],在MTB临床分离株中,tlya突变并不常见[8,10,22],而且CPM敏感的临床分离株中也存在该基因突变[22-23,25],表明tlya并非MTB临床分离株对CPM耐药的敏感标记[22]。多聚磷酸盐/ATP依赖性NAD激酶(Polyphophate /adenosine triphosphate (ATP)-dependent NAD kinase,ppnK)是MTB生长的必需基因[26],有研究通过构建耻垢分枝杆菌转座子突变体文库,分离到一个抗CPM的突变体c4,测序发现c4在tlya和ppnk之间重叠区发生了A到G的替换,提示tlya和ppnk重叠区突变可能与CPM耐药有关,但其机制有待进一步阐明[27]。
2.2rrs基因rrs基因编码16S rRNA,该基因突变导致CPM作用靶点的改变,使其失去对MTB的作用,从而产生耐药[8-9]。其中最常见的是A1401G突变[13,28],但有些研究用大肠杆菌16S rRNA基因中的1408位点表示[29]。据计算,A1401G突变占CPM耐药比例的76%[30],其突变可导致CPM与卡那霉素或阿米卡星交叉耐药[22-24,28,31],但该突变的临床耐药株对CPM的最小抑菌浓度(Minimum Inhibitory Concentration, MIC)存在较大差异(表1),且该突变也存在于CPM敏感株中[9,12,22,31]。对于MIC值的差异,推测有以下两点原因:1.不同的药物敏感试验(Drug Susceptibility Testing, DST)方法和标准之间判定的DST结果差异较大,可能出现错误的易感结果[32];在CPM耐药性的检测中,MGIT960系统DST结果与A1401G突变的一致性更好,高于琼脂比例法的检测[32-33]。2.可能存在第二个位点或基因表达状态的改变,从而产生协同或拮抗作用[9,34],如tlya表达增加时,该突变的抗性降低[35]。rrs基因的其他突变位点见表1。据报道C1402T和G1484T位点突变普遍导致MTB对CPM高度耐药,且与交叉耐药有关[28,31]。C517T突变导致CPM中度至高度耐药(MIC,20~80 μg/mL)[8]。最近,在rrs基因中发现一个新的突变G878A,含G878A的突变株其CPM的MIC为8~64 μg/mL,该作者推测G878A突变是CPM耐药的新机制,并提出在新分子检测中加入G878A可提高CPM耐药检测的灵敏度,同时该研究发现这一突变与欧美x3型MTB有关[15]。
2.3eis启动子 研究发现eis突变与低水平的CPM耐药有关[13-15],也可导致CPM交叉耐药[14],目前报道的突变位点见表1。G10A突变相对常见,但已有研究发现在敏感株中也存在该突变[22],推测eis和CPM之间的相互作用可能受到MTB个体化因素的影响[13]。
2.4其他 除上述3个CPM耐药相关基因外,研究发现在MTB和大肠杆菌中23S rRNArrl基因A1916的缺失导致CPM耐药[11],随后发现,在嗜热杆菌(Thermusthermophilus)中23S rRNA螺旋H69的3个突变(A1913U 、mU1915G △mU1915)也引起CPM的耐药[16]。此外,rpsL基因突变也可引起CPM耐药(表1),且CPM的耐药率在不同地区存在差异[36-37]。
表1 CPM相关基因突变频率
Tab.1 Mutation frequency of capreomycin related gene
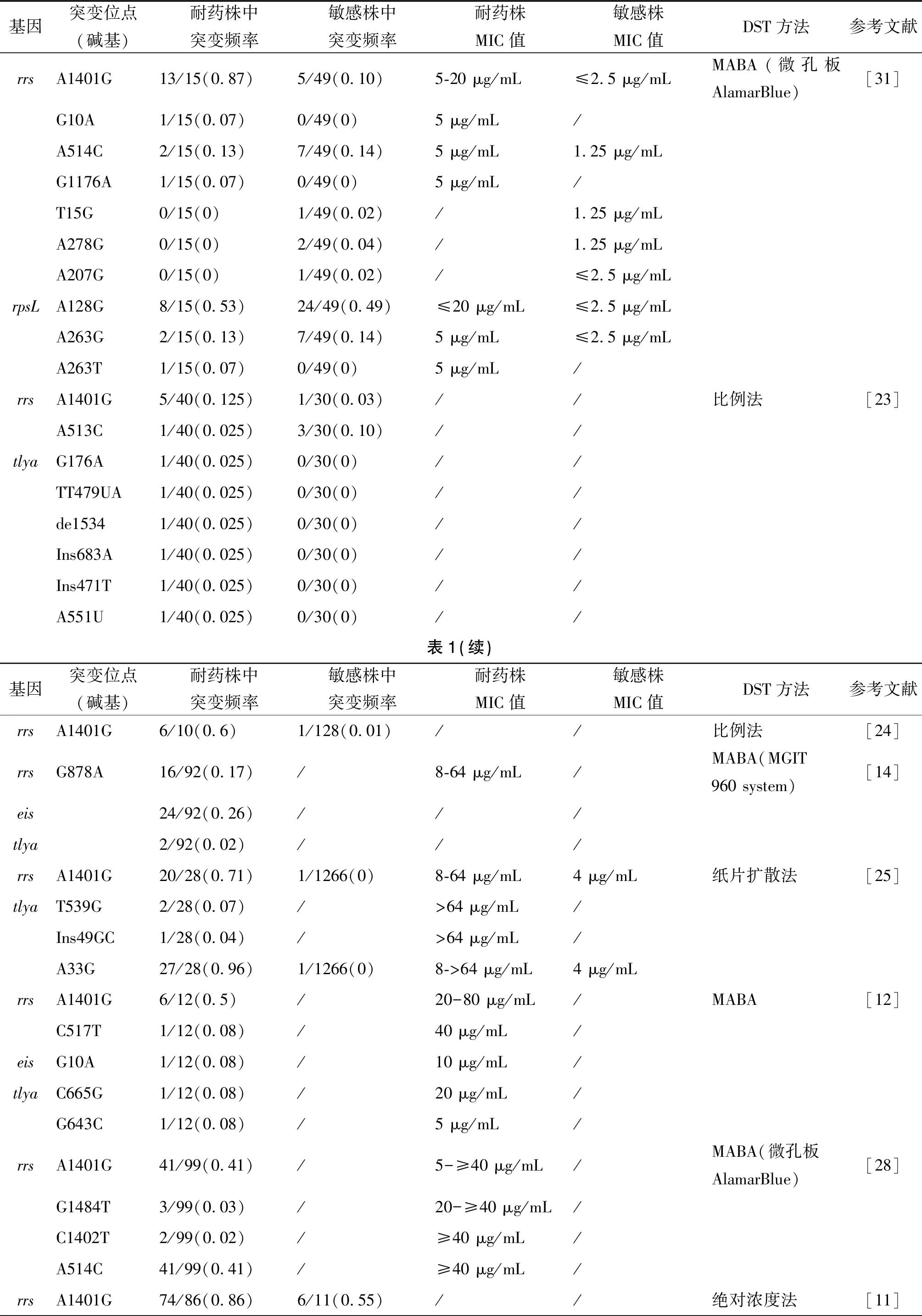
基因突变位点(碱基)耐药株中突变频率敏感株中突变频率耐药株MIC值敏感株MIC值DST方法参考文献rrsA1401G13/15(0.87)5/49(0.10)5-20 μg/mL≤2.5 μg/mLMABA(微孔板AlamarBlue)[31]G10A1/15(0.07)0/49(0)5 μg/mL/A514C2/15(0.13)7/49(0.14)5 μg/mL1.25 μg/mLG1176A1/15(0.07)0/49(0)5 μg/mL/T15G0/15(0)1/49(0.02)/1.25 μg/mLA278G0/15(0)2/49(0.04)/1.25 μg/mLA207G0/15(0)1/49(0.02)/≤2.5 μg/mLrpsLA128G8/15(0.53)24/49(0.49)≤20 μg/mL≤2.5 μg/mLA263G2/15(0.13)7/49(0.14)5 μg/mL≤2.5 μg/mLA263T1/15(0.07)0/49(0)5 μg/mL/rrsA1401G5/40(0.125)1/30(0.03)//比例法[23]A513C 1/40(0.025)3/30(0.10)//tlyaG176A1/40(0.025)0/30(0)//TT479UA1/40(0.025)0/30(0)//de15341/40(0.025)0/30(0)//Ins683A1/40(0.025)0/30(0)//Ins471T1/40(0.025)0/30(0)//A551U1/40(0.025)0/30(0)//表1(续)基因突变位点(碱基)耐药株中突变频率敏感株中突变频率耐药株MIC值敏感株MIC值DST方法参考文献rrsA1401G6/10(0.6)1/128(0.01)//比例法[24]rrsG878A16/92(0.17)/8-64 μg/mL/MABA(MGIT 960 system)[14]eis24/92(0.26)///tlya2/92(0.02)///rrsA1401G20/28(0.71)1/1266(0)8-64 μg/mL4 μg/mL纸片扩散法[25]tlyaT539G2/28(0.07)/>64 μg/mL/Ins49GC1/28(0.04)/>64 μg/mL/A33G27/28(0.96)1/1266(0)8->64 μg/mL4 μg/mLrrsA1401G6/12(0.5)/20-80 μg/mL/MABA[12]C517T1/12(0.08)/40 μg/mL/eisG10A1/12(0.08)/10 μg/mL/tlyaC665G1/12(0.08)/20 μg/mL/G643C1/12(0.08)/5 μg/mL/rrsA1401G41/99(0.41)/5-≥40 μg/mL/MABA(微孔板AlamarBlue)[28]G1484T3/99(0.03)/20-≥40 μg/mL/C1402T2/99(0.02)/≥40 μg/mL/A514C41/99(0.41)/≥40 μg/mL/rrsA1401G74/86(0.86)6/11(0.55)//绝对浓度法[11]
注: / 表示原文无数据
3 展 望
CPM是治疗耐多药结核病的二线药物,目前对其作用机制及耐药机制的研究已取得一定的进展。大多数的研究认为CPM主要通过抑制蛋白质合成、影响细胞壁的功能等发挥其抗结核分枝杆菌作用,其耐药性的产生主要与rrs、tlya、eis等基因的突变有关,但其具体的作用及耐药机制还有待进一步研究,如核糖体蛋白L12和L10、tlya和ppnk重叠区的突变是通过何种途径影响CPM的抗菌活性目前仍不清楚。本文对CPM相关基因的突变频率进行了统计,发现其耐药所涉及的相关基因其突变范围较广泛、突变频率较低、MIC值变化较大,目前还难以确定CPM耐药性检测的敏感分子标记,且不同的DST检测方法之间存在差异,目前尚缺乏统一标准,具体检测方法还有待进一步统一;此外,已有报道在CPM耐药株中未检测到当前发现的突变位点,提示可能存在其他的耐药机制,需要进行更深入的研究以验证当前的研究结果,并探索其作用的新机制。对CPM的作用及耐药机制的深入研究可能为耐药基因检测技术的发展和指导临床合理用药提供新的思路和方法,为结核病及耐药结核病的诊断提供更科学有力的支撑。
利益冲突:无
猜你喜欢
山东医药(2022年6期)2023-01-06
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22
生物化学与生物物理进展(2022年9期)2022-09-22
保健医苑(2022年5期)2022-06-10
医学概论(2022年4期)2022-04-24
大众科学(2022年3期)2022-04-09
中国生殖健康(2020年2期)2021-01-18
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
