
Mutation identification of PAX6 and prenatal diagnosis in a Chinese family with Aniridia and gestational diabetes
2019-09-17 05:40:08ShiQiDongSuFangDong2ChenQiao3BoHu4FangZheng5MingYan
国际眼科杂志 2019年9期
Shi-Qi Dong*, Su-Fang Dong2*, Chen Qiao3, Bo Hu4, Fang Zheng5, Ming Yan
Abstract
•KEYWORDS:aniridia; mutation; PAX6; prenatal diagnosis
INTRODUCTION
Aniridia (AN) is a congenital, bilateral, panocular disorder characterized by complete absence of the iris or partial iris hypoplasia associated with reduced visual acuity, nystagmus, and foveal hypoplasia. It is not only an isolated defect in iris development, but it is associated with macular and optic nerve hypoplasia, cataract, glaucoma, corneal changes and nystagmus[1]. Two-thirds of aniridia cases are inherited in an autosomal dominant trait with variable expressivity, and the other cases are sporadic[2].
It is notable that aniridia usually has been thought caused by mutation of the paired box 6 (PAX6) gene which is a member of the paired box gene family, locates on chromosome 11p13 and encodes a transcriptional regulator involved in oculogenesis, and other developmental processes, such as pancreatic, pituitary, central nervous system[3-6]. The mutations in PAX6 caused aniridia are scattered throughout the whole gene and the vast majority of those reported so far are nonsense mutations, frame-shift mutations, or splicing errors that are predicted to create a premature truncation protein, leading to haploinsufficiency[1]. Recently, the relationship between the PAX6 mutation and the differentiation and function of islet cells enters into the scope of hot research. There were many reports of impaired glucose tolerance or diabetes in PAX6 mutation carriers with aniridia[7-8].
It could be argued that prenatal testing should only be offered when termination or corrective treatment is possible. Follow the guidelines of prenatal diagnosis application in China,and prenatal diagnosis could be performed for the reason of preparing in the aspect of mentally, financial and medical supply when one of the couples carried hereditary diseases[9]. During childhood, it is important for taking effective measures to prevent further visual impairment, such as to correct the refractive errors using spectacles.
Total number of unique DNA variants reported in the human PAX6 allelic variant database is 357. In this study, we performed clinical diagnosis, genetics analysis and prenatal diagnosis of a Chinese family with aniridia. A heterozygous deletion mutation in PAX6 was identified in our study.
SUBJECTS AND METHODS
SubjectRecruitmentandClinicalExaminationA Chinese AN pedigree was recruited in Zhongnan Hospital of Wuhan University (Wuhan, China). Written informed consent was obtained from all participating adults (Figure 1). The participating affected individuals underwent clinical examination. And the pregnant proband received a complete ophthalmic evaluation including visual acuity, the ocular slit-lamp examination, and blood glucose assay using oral glucose tolerance test. This research was approved by Zhongnan Hospital Research Ethics Committee and followed the tenets of the Declaration of Helsinki.
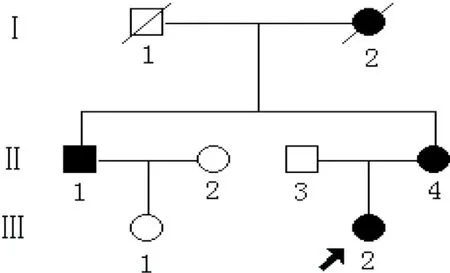
Figure1AChinesepedigreewithaniridiaandcataractThe proband is marked with a black arrow.
SampleCollectionandGeneticsAnalysisThe peripheral venous blood samples were collected for genomic DNA extractionviathe standard protocol and procedures, and genomic DNA samples were stored at -20°C before using. All coding exons in the known candidate gene PAX6, as well as its flanking regions, were amplified by PCR method. The PCR productions were sequenced by ABI 3130 genetic analyzer (Life Technologies Corporation, CA, USA) after purification. The sequencing results were compared with the reference sequence in the NCBI database.
PrenatalDiagnosisAmniocentesis was executed on the affected female at 18wk of gestation by an obstetrician under ultrasonic monitoring, and all the procedures were normatively operated. Approximately 20 mL amniotic fluid (AF) was drawn which visually appearing to be untainted with blood. Then AFC were cultured using Chang Medium©insitu(Irvine Scientific, CA, USA) under 5% CO2in a 37℃ incubator. Another written informed consent was obtained from this patient to use the AF for genetic analysis. Both the obstetrician and the doctor for genetic analysis had the qualification for prenatal diagnosis.
AFC were washed three times with PBS and trypsinogen with 0.25% Gibco©Trypsin (Invitrogen Corporation, CA, America) for 5min at 37℃ after ten days’ culture. Then genomic DNA was isolated from AFC through the standard phenol/chloroform extraction. The PCR and sequencing were performed with the aforementioned methods in genetic analysis.
RESULTS
ClinicalDiagnosisThe affected in the family associated with AN who had typical clinical features such as completely bilateral aniridia, congenital nystagmus and severe congenital cataract. Applanation tonometry revealed normal intraocular pressure in both eyes of the proband, and the width of the corneas was 10.5 mm in both eyes. The lens had partial opacity (Figure 2). The visual acuity measured by the logarithm of the minimum angle of resolution (LogMAR) was 2.0 in both eyes. Oral glucose tolerance test showed a normal fasting blood glucose level (4.9 mmol/L) and 1h glucose level (9.8 mmol/L) whereas the concentration of 2-hour blood glucose was 8.8 mmol/L. Therefore, the affected female was diagnosed as suffering from gestational diabetes.
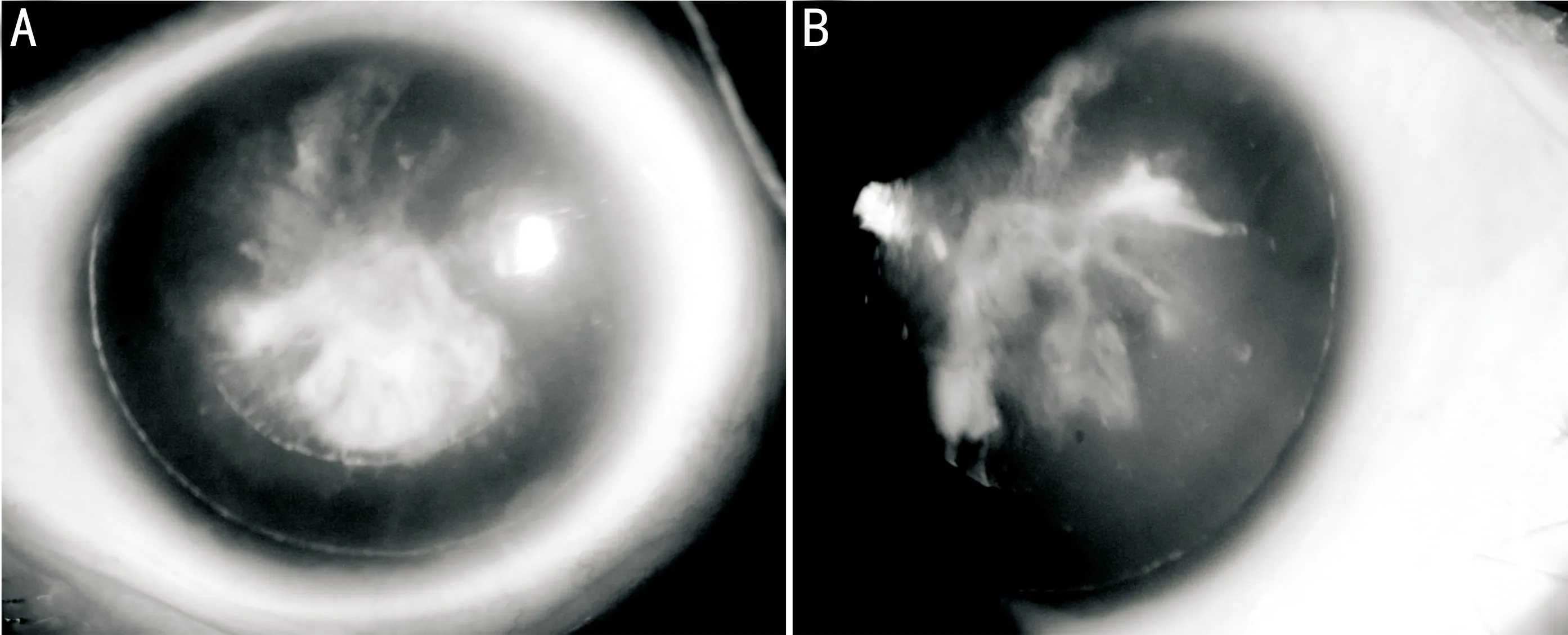
Figure2Slit-lampphotosoftheocularanteriorsegmentoftheproband.Theproband(II-1)exhibitsaniridiawithoutirisremnantsandcataract(A: The right eye; B: The left eye).

Figure3ChromatogramsofmutationscreeninginPAX6A: Showing heterozygous deletion mutation in PAX6 in an affected individual with aniridia; B: The AFC genetic analysis showed the fetus carried the same mutation.
MutationIdentificationTo identify the causative mutation in this pedigree, we screened the whole encoding regions of PAX6 in all recruited family members by PCR-based direct DNA sequencing. A heterozygous deletion mutation was identified in the patients that a 17bp (c.113_129delGGCCGTGCGACATTTCC) was deleted in exon 5 of PAX6 (Figure 3). It resulted in a frame-shifting deletion mutation which was predicted to lead to a premature termination codon (p.Arg38ProfsTer12). The mutation was not found in normal family members.
PrenatalDiagnosisWith exclusions of any prescient contaminations such as discarding the first 2 mL amniotic fluid after puncture, washing cultured AFC before trypsinization, setting up negative control of PCR and so on, it suggested that DNA from AFC carried the same heterozygous deletion mutation like the proband (Figure 3). It indicated that the fetus would be suffering from AN. And the postpartum follow-up confirmed the result that the infant was affected with AN and congenital cataract.
DISCUSSION
We describe an AN family with three affected and four unaffected members available for examination. The affected family members had a total absence of the iris, congenital nystagmus, seriously congenital cataract, and impaired visual acuity. The results of genetics analysis confirmed the presence of PAX6 mutation that a 17bp deletion mutation (c.113_129delGGCCGTGCGACATTTCC) located in exon 5 led to a premature termination codon (p.Arg38ProfsTer12). It is a rare mutation although it has been reported in another Chinese family[10]. Genetics analysis of AFC indicated the fetus carrying the same mutation like the mother and the subsequently postpartum follow-up confirmed the result of the prenatal diagnosis that the infant was affected with aniridia and congenital cataract. It is noteworthy that the affected female underwent prenatal diagnosis was also suffering from gestational diabetes.
PAX6 is expressed in the developing eye, multiple brain regions, olfactory bulb, spinal cord, gut and pancreas[11-12]. It is known to play a crucial role in the development of the retina, lens, cornea and iris[13-14]. Although there are patients with AN caused by PAX6 cis-regulatory element (SIMO) that resides in an intron of the adjacent elongator protein complex 4 (ELP4) gene[15-16], AN is usually caused by heterozygous mutation in the PAX6 gene through the effect of its haploinsufficiency in most patients. Human PAX6 encodes a 422 amino acid transcriptional regulator, contains two DNA-binding domains, a bipartite paired domain (PD) and a paired-type homeobox domain (HD), separated by a linker region for its binding activity. The PD domain is composed of the N-terminal subdomain (NTS) and the C-terminal subdomain (CTS)[17]. In our research, the delete mutation lied on the NTS region which is highly conserved among members of the PAX family of genes and plays a vital role in binding with the DNA[18]. Actually, most mutations in PAX6 associated with AN occur in the NTS where the DNA-binding ability may be altered. The p.Arg38ProfsTer12 mutation in PAX6 produced a premature termination codon (TAA) and thereby generates a truncated protein that lacks part of the PD and all of the other domains. We reviewed the archived mutations in the PAX6 Allelic Variant Database and found that over 90% mutations in PAX6 led to the premature truncation of the protein just like our study demonstrated. These mutations are predicted to disrupt transcription or translation and likely to be pathological mutations.
As noted earlier, PAX6 plays an essential role in islet cell differentiation andfunction[3,19]. Disruption of the PAX6 gene in mice causes a marked effect in glucose homeostasis and decrease the expression of important islet cell markers[20]. PAX6 mutation carriers with aniridia were found to have impaired glucose tolerance or diabetes in previous studies, and mutant PAX6 downregulates prohormone convertase two expressions might be contributing to the underlying mechanism[7,21]. In our research, the II1 with aniridia is also diabetic, and the affected female III2 has gestational diabetes, but we could not affirm the relationship between this PAX6 mutation and islet cell development since it needs further experimental analysis, such as testing blood glucose to judge whether III2 had impaired glucose tolerance after parturition.
It is indisputable that there is a close relationship between aniridia and PAX6 mutations. It means that prenatal testing can theoretically be offered to most people. AN is not a life-threatening condition but does severely affect sight as well as companies with many complications such as glaucoma and cataract. Many researchers reported the mutations in PAX6 are also associated with the abnormality of the central nervous system and glucose intolerance and so on[7,22-23]. Therefore, it will puzzle parents for children’s healthy growth. Based on family requesting advice about prenatal diagnosis and following indication of management measures of prenatal diagnosis in Chinese[9], we performed the testing through the amniocentesis. The result suggested the fetus carried the same mutation like the mother. After the couple was informed of the results of the genetic analysis, they expressed gratitude to us and insisted that they wanted the child, even though the baby had an iris-free and congenital cataract. Then the proband couple began to prepare for the birth of the baby, especially the mental education problem.
In summary, this study identified a deletion mutation (p.Arg38ProfsTer12) of PAX6 in a Chinese family with AN. Since the AN patient is accompanied by corneal degeneration, eyeball horizontal tremors, and cataracts, this research is more valuable for genetic counseling and prenatal diagnosis.
