
QuEChERS与液相色谱-串联质谱联用检测中药、调味品及外敷药物中去甲乌药碱的含量
2019-09-13 08:57汤一铸赵晓亚郭少飞吴建安
色谱 2019年10期
王 晗, 汤一铸, 赵晓亚, 叶 诚, 郭少飞, 罗 静,熊 露, 曹 维, 吴建安, 王 鹏*
(1. 武汉海关技术中心, 湖北 武汉 430050; 2. 湖北省体育科学研究院, 湖北 武汉 430205)
去甲乌药碱是许多天然植物中存在的β-2激动剂类药物,具有刺激β-肾上腺素受体、舒张血管的作用,因此在临床上可被用作强心剂[1-4]。自2017年起,去甲乌药碱就被国际反兴奋剂组织(WADA)列为S3类(β-2激动剂类)违禁物质,要求不得检出。去甲乌药碱主要存在于南天竹、附子、细辛、番荔枝、莲等天然植物中[5,6],这些植物正是中药材或日常饮食调味品的组成部分,此外,其也可能存在于以这些天然药物作为原料的外用药物中。因此对日常饮食及所使用的中药药物中去甲乌药碱的检测有利于避免运动员因误食误用而导致的兴奋剂阳性检出。基于此,必须建立针对日常饮食及治疗中可能涉及的中药、调味品和外敷药物的高灵敏、可靠的去甲乌药碱检测方法。
去甲乌药碱的常见检测手段包括液相色谱-紫外光谱法、液相色谱-荧光法[7]、液相色谱-电化学检测法[8]和液相色谱-串联质谱检测法[9,10]等,其中液相色谱-串联质谱法具有灵敏度高、选择性好的优点,已被应用于运动员血清、尿样[10]以及保健品[9]、中药[11]中去甲乌药碱的检测。
由于中药、调味品及外敷药物的基质复杂多样,去甲乌药碱在样品中的存在含量通常较低,且对中药、调味品及外敷药物中去甲乌药碱需要进行大量的筛查,因此亟须建立快速、灵敏、可批处理的前处理方法对样品中的去甲乌药碱进行分析。传统的样品前处理方法包括液相萃取技术和固相萃取技术等,其中,液相萃取技术具有操作简单、萃取速度快的优点,但样品基质效应影响严重;固相萃取技术具有萃取材料种类丰富、样品基质效应影响小的优点,但操作较为繁琐。近年来,QuEChERS方法作为一种新型样品前处理手段获得了广泛的关注,由于其具有操作简便、可批处理、分析速度快等优点,已被广泛应用于食品[12]、环境样品[13]中的农药残留[14]、兽药残留[15]、生物毒素[16]等目标分析物的检测。
本工作旨在将QuEChERS样品前处理技术与液相色谱-串联质谱联用,建立针对中药、调味品及外敷药物中去甲乌药碱的简便、可靠、可批处理、高灵敏检测方法。
1 实验部分
1.1 仪器、药品、试剂及实际样品
岛津LC-20ADXR型液相色谱仪(Shimadzu,日本), QTRAP6500型串联质谱仪(SCIEX,美国), MS型分析天平(Mettler Toledo,上海), 2-16PK型低温离心机(Sigma,德国), SA300型振荡器(Yamato,日本), MS2型涡旋混合器(IKA,德国), TurboVap LV型氮吹仪(Biotage,瑞典)。去甲乌药碱(纯度99.0%)、非诺特罗(内标,纯度99.0%)购自天津阿尔塔科技有限公司,甲醇、乙醇、甲酸为色谱纯试剂(LiChrosolv, Merck,德国)。QuEChERS吸附剂乙二胺-N-丙基硅烷(PSA)、十八烷基键合硅胶(C18)及石墨化炭黑(GCB)均购自CNW Technology公司(上海)。
本工作建立的方法应用于13种中药、4种调味料及1种藏药贴膏中的去甲乌药碱含量的检测,实际样品包括市售的干荷叶、茯苓、黄芪、当归、水栀子、杏仁、田七、川芎、白芍、干莲子、山药、玉竹、淮山、辣椒、茴香、花椒、桂皮和由湖北省体育科学研究院提供的藏药骨痛贴膏。空白样品采用市售的大米样品。
1.2 溶液配制
分别称取去甲乌药碱和非诺特罗标准品(精确至0.01 mg),配制成质量浓度为1.0 g/L的标准溶液,于-18 ℃下避光保存。在实验过程中精确吸取适量标准溶液用0.1%(v/v)甲酸-乙醇溶液逐级稀释至所需浓度。
1.3 样品前处理
取适量实际样品,用粉碎机粉碎后过筛备用,粉碎后若不均匀,可用瓷研钵研磨至均匀后过筛备用;藏药贴膏用剪刀剪开后,取贴膏内药粉。
提取及净化:取固体样品约2.0 g(精确至0.001 g)于15 mL离心管中,加入5 mL含0.5%(v/v)甲酸的乙醇溶液、50 μL 1.0 mg/L内标(非诺特罗)溶液,涡旋混合均匀后于振荡器上振荡提取30 min,离心10 min(8 000 r/min),取上清液1 mL于1.5 mL离心管中;加入50 mg PSA、50 mg C18及7.5 mg GCB,涡旋处理1 min后离心5 min(8 000 r/min),取上清液用0.22 μm的微孔滤膜过滤,滤液供LC-MS/MS测定,若样品溶液浓度过高,用0.5%(v/v)甲酸-乙醇溶液稀释后进样。实验中所有的样品均设置3个平行组。
1.4 色谱及质谱条件
Waters atlantis T3色谱柱(100 mm×3 mm, 3 μm);流速:0.40 mL/min;柱温:30 ℃;进样量:5 μL。流动相:A相为含0.1%(v/v)甲酸的0.5 mmol/L乙酸铵水溶液;B相为含0.1%(v/v)甲酸的甲醇溶液。梯度洗脱程序如下:0~2.0 min, 5%B; 2.0~2.4 min, 5%B~50%B; 2.4~3.0 min, 50%B; 3.0~3.4 min, 50%B~95%B; 3.4~4.0 min, 95%B; 4.0~4.1 min, 95%B~5%B; 4.1~7.0 min, 5%B。
离子化模式:ESI+;电喷雾电压(IS): 5 500 V;气帘气为241 325 Pa;雾化气压力为344 800 Pa;辅助气压力为413 700 Pa;检测方式:多反应监测;去甲乌药碱定性离子对为m/z272.0/255.0、272.0/161.0,定量离子对为m/z272.0/107.0,去簇电压(DP)为40 V,碰撞能量(CE)分别为20、25、30 eV。内标检测离子对为m/z304.2/135.3。
2 结果与讨论
2.1 液相色谱-串联质谱检测条件优化
将1.0 mg/L去甲乌药碱溶液以7 μL/min的速度引入质谱,分别于正离子模式和负离子模式进行母离子扫描,得到去甲乌药碱的母离子为m/z272.0;再对母离子进行二级质谱分析,选择丰度较强的3对子离子,其中定量子离子为m/z272.0/107.0,定性子离子为m/z272.0/255.0、272.0/161.0;最后优化去簇电压(DP)和碰撞能量(CE),最终选择去簇电压(DP)为40 V,离子对m/z272.0/255.0、272.0/161.0、272.0/107.0的碰撞能量(CE)分别为20、25、30 eV。内标非诺特罗的优化同上进行。
去甲乌药碱色谱检测条件的优化是在文献[11]的基础上进行微调完成的。采用花椒和桂皮混合基质提取液加标100 μg/L作为模拟样品,以检测离子对m/z272.0/255.0、272.0/161.0、272.0/107.0的色谱分离情况为标准,考察不同流动相梯度洗脱条件下去甲乌药碱与基质的分离情况。结果表明,按照文献[11]中的洗脱程序,在从5%B直接提升至95%B的洗脱过程中,混合基质提取液中有干扰峰与去甲乌药碱色谱峰无法基线分离,因此将洗脱程序修改为从5%B提升至50%B后维持0.6 min,再从50%B提升至95%B,最终确定了1.4中的色谱分离条件。
2.2 样品提取条件的选择
在样品提取条件优化阶段,向空白样品中加入去甲乌药碱标准溶液至样品中去甲乌药碱含量为20 μg/kg作为模拟样品。提取条件的选择以去甲乌药碱回收率(提取效率)的高低作为判断标准。去甲乌药碱是天然植物中存在的苄基异喹啉类生物碱[17],其油水分配系数(logP)为2.23, 解离常数(pKa)为9.72,由于其是具有较强极性的生物碱,因此适合采用含酸的极性溶剂进行提取。本工作中首先采用5 mL含有盐酸的水溶液、甲醇和乙醇作为提取溶剂进行考察,结果如图1所示,可知乙醇的提取效率最高。

图 1 采用不同提取溶剂时去甲乌药碱的提取效率(n=3)Fig. 1 Extraction efficiencies of higenamine with different extraction solvents (n=3)
此后,采用5 mL含有0、0.1%(v/v)、0.2%(v/v)、0.5%(v/v)、1.0%(v/v)甲酸的乙醇作为提取溶剂,结果见图2。结果表明,去甲乌药碱的提取效率会随着甲酸的加入而有所提高,但当甲酸含量达到0.2%(v/v)以后,就基本不再变化,因此在后续实验中,采用含0.5%(v/v)甲酸的乙醇溶液作为去甲乌药碱的提取溶剂。

图 2 采用含不同体积分数甲酸的乙醇溶液提取时去甲乌药碱的提取效率(n=3)Fig. 2 Extraction efficiencies of higenamine with ethanol containing different volume percentages of formic acid (n=3)
由图1的结果可知,在处理实际样品时,提取溶剂的极性会极大地影响去甲乌药碱的提取,由于水与乙醇可互溶,样品含水量的不同很可能影响提取体系的极性从而影响去甲乌药碱的提取效率。以干燥大米样品为空白基质,向样品中加适量蒸馏水模拟含水量为0、20%(质量分数)、33%(质量分数)和50%(质量分数)的实际样品,采用5 mL含0.5%(v/v)甲酸的乙醇溶液进行提取,考察去甲乌药碱的提取效率。结果表明,样品中含水量的上升会引起去甲乌药碱提取效率的降低,当样品不含水时,去甲乌药碱的提取效率在70%左右,而样品含水量上升至20%(质量分数)时,提取效率就会下降至35%左右。考虑到中药和调味品一般都是干样,在提取过程中,选择不水化直接采用含0.5%(v/v)甲酸的乙醇溶液提取。
2.3 其他提取条件的选择
采用加标空白样品及花椒样品比较了在振荡提取前不浸泡、浸泡0.5 h以及浸泡1 h的提取效率,其提取效率分别为72.8%、68.1%和75.0%,结果表明浸泡与否对去甲乌药碱的提取没有影响,因此在后续实验中不浸泡样品,直接提取。
采用加标空白样品及花椒样品考察了振荡时间20、30、45和60 min的提取效率,结果表明,振荡时间对去甲乌药碱的提取没有明显的影响,因此,选择30 min作为振荡提取时间。
2.4 QuEChERS净化条件的选择
PSA、C18和GCB是QuEChERS净化过程中常用的净化剂,为了保证在样品净化过程中去甲乌药碱不会被吸附剂吸附,本工作以加标空白大米样品中去甲乌药碱的回收率为考察指标,采用三水平三因素的正交试验对3种吸附剂的使用量进行了考察,三水平三因素的正交试验条件及结果列于表1。由表1可知,3种吸附剂对去甲乌药碱回收率的影响水平为PSA>C18≈GCB,其中,PSA使用量越高,去甲乌药碱的回收率越低;而C18和GCB的加入对去甲乌药碱的回收率几乎没有影响;考虑到随着GCB加入量的增加,可以看到样品颜色明显变浅,表明更多色素被吸附剂吸附,但由于GCB为广谱型吸附剂,当样品提取液基质简单时,易造成对去甲乌药碱的吸附,因此在后续实验中,采用50 mg PSA、50 mg C18及7.5 mg GCB作为吸附剂对样品进行净化。
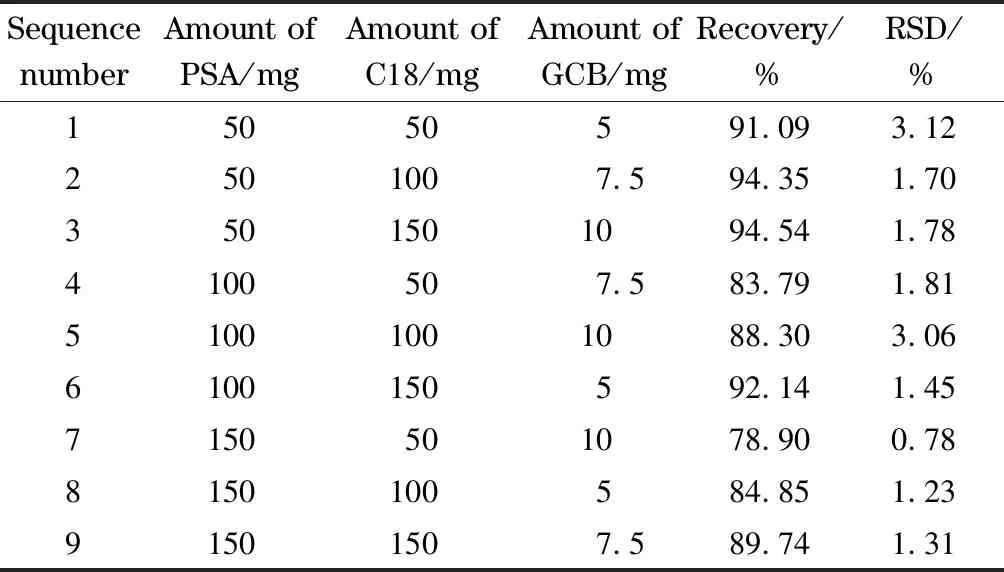
表 1 正交试验条件及去甲乌药碱回收率的结果(n=3)
2.5 方法分析性能的考察
考虑到实际样品的基质种类复杂,对去甲乌药碱的提取和净化会造成较大的影响,因此本工作采用内标法对去甲乌药碱进行检测。选择与去甲乌药碱性质相似的动物源性β-2激动剂非诺特罗(logP值为2.06,pKa为9.25)作为内标物质。
为了验证内标法的必要性,考察了不同样品的基质效应:取3种不同空白样品(大米、辣椒、水栀子)的提取净化液加标至去甲乌药碱质量浓度为100 μg/L,取0.5%(v/v)甲酸的乙醇溶液直接配制100 μg/L的去甲乌药碱溶液,将4份溶液分别引入液相色谱-串联质谱仪检测,结果表明相比于以0.5%(v/v)甲酸的乙醇溶液配制的去甲乌药碱溶液,大米、辣椒和水栀子3种样品的提取净化液加标溶液中去甲乌药碱的峰面积为92.9%、27.7%和18.2%,这表明样品基质的不同对去甲乌药碱的检测具有重大影响,因此在本工作中采用内标进行定量具有必要性。
在最优的实验条件下,对方法的分析性能进行了考察,方法的检出限(去甲乌药碱响应值为噪声3倍时的含量)为0.03 ng/g,线性范围为0.10~100 ng/g,线性方程y=0.307 8x-0.002 4(r2=0.993 3)(x为去甲乌药碱与内标含量比,y为去甲乌药碱与内标峰面积比)。方法的精密度为4.33% (0.50 ng/g,n=7),图3所示为检出限水平的样品色谱图。根据WADA 2019年对尿液中去甲乌药碱的检测限量要求(10 μg/L)[18],本方法具有更高的灵敏度,完全可以满足运动员日常饮食和外敷用药中去甲乌药碱的检测需求。

图 3 去甲乌药碱(0.03 ng/g)和内标(25 ng/g)的提取离子色谱图Fig. 3 Chromatograms of higenamine (0.03 ng/g) and internal standard (25 ng/g)
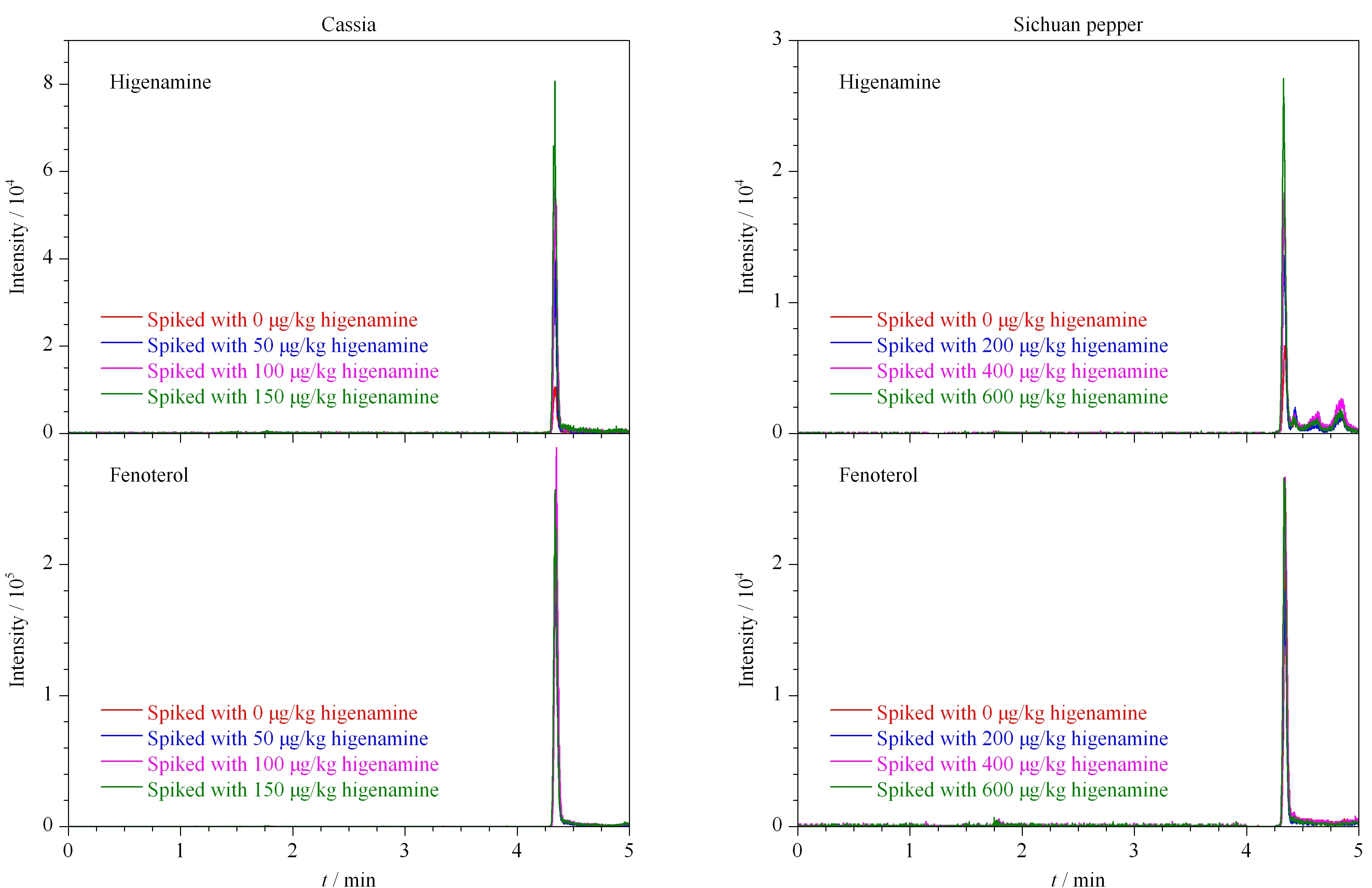
图 4 花椒和桂皮样品及加标样品中去甲乌药碱和内标的提取离子色谱图Fig. 4 Chromatograms of higenamine and internal standard in Cassia and Sichuan pepper samples, and their spiked samples
2.6 实际样品分析及加标回收率的考察
采用所建立的方法对实际样品中去甲乌药碱的含量进行了检测,其中,干荷叶、干莲子、山药、玉竹、淮山、花椒、桂皮和藏药骨痛贴膏中均能检出去甲乌药碱,其含量见表2。

表 2 实际样品中去甲乌药碱的含量(n=3)
采用加标回收试验验证方法的准确性,对花椒、桂皮两种实际样品进行加标,其样品及样品加标提取离子色谱图见图4,加标回收率结果见表3。由结果可知,去甲乌药碱的加标回收率为92.6%~109.8%,表明本方法准确性良好。

表 3 去甲乌药碱在实际样品中的加标回收率及RSD(n=3)
3 结论
本工作建立了基于QuEChERS前处理技术与LC-MS/MS联用检测去甲乌药碱的新方法。该方法具有灵敏度高、操作简便、分析迅速的优点,适用于中药、调味料和外敷用药中去甲乌药碱含量的测定。采用所建立的方法对多种中药及调味品进行检测,在干荷叶、干莲子、山药、玉竹、淮山、花椒、桂皮和藏药骨痛贴膏中检测到了去甲乌药碱,采用加标回收试验验证方法的准确性,回收率为92.6%~109.8%,表明本方法具有较好的准确性,适用于植物源性食品,特别是中药、调味料和外敷用药中去甲乌药碱的快速、批量筛查检测。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
食品研究与开发(2021年22期)2021-12-06
延安大学学报(自然科学版)(2020年4期)2021-01-15
中成药(2018年7期)2018-08-04
南方农业·上旬(2018年7期)2018-05-14
中成药(2017年10期)2017-11-16
中国卫生标准管理(2015年2期)2016-01-14
医学研究杂志(2015年4期)2015-06-10
浙江中西医结合杂志(2015年12期)2015-06-06
中国民族民间医药·下半月(2014年2期)2014-09-26
