
超高效液相色谱-串联质谱法快速测定保健食品中大豆异黄酮含量
2019-09-11 07:55胡珀,金华
食品工业科技 2019年13期
胡 珀,金 华
(淮安市食品药品检验所,江苏淮安 223001)
大豆异黄酮是大豆中的一类次生代谢产物,属黄酮类中的异黄酮多酚化合物[1]。目前发现的大豆异黄酮共有12种,包括游离型苷元和相应糖苷[2]。大豆异黄酮是天然的优质抗氧化剂,具有很强的清除羟自由基(-OH)的能力;具有防癌抗肿瘤、降低胆固醇、预防骨质疏松和心血管疾病、延缓衰老、改善妇女更年期综合症等多种生理功能[3-6]。因此,以大豆异黄酮为主要活性成分的保健食品的开发和应用受到广泛关注。其中大豆苷(Daidzin)、大豆黄苷(Glycitin)、染料木苷(Genistin)、大豆素(Daidzein)、大豆黄素(Glycitein)以及染料木素(Genistein)六种大豆异黄酮单体是大豆中异黄酮的主要组分。
目前,保健食品市场存在生产低水平重复、价位较高、夸大产品功效的现象。因此,应该提高保健食品功效成分的检测分析水平[7]。保健食品中大豆异黄酮含量的传统测定方法有紫外分光光度法(UV),该法具有方法简便、重现性好等优点,但特异性较差,无法判断样品中各组分的构成情况[8];薄层扫描法(TLCS),该法具有取样量少、分离效果好等优点,能较好地进行定性分析,但定量检测精度不高,人为误差较大[9];气相色谱法(GC)法具有进样量少、特异性高等优点,但在测定时需制备衍生物,步骤较多,耗时长,从而限制了该法的推广应用[8];酶联免疫吸附法(ELISA)具有检测灵敏度高、检测速度快的优点,多用于生物体血液及尿样中大豆异黄酮的检测[10];高效液相色谱法(HPLC)主要是参照GB/T 23788-2009《保健食品中大豆异黄酮的测定方法高效液相色谱法》[11],该分析方法具有自动化程度高、测定结果准确、重现性好等优点,但仪器运行时间较长,需要60 min,不适合研发检测,另外,待检样品基质复杂,大豆异黄酮含量低,不易检出,长期直接进样,易污染色谱柱。
本课题采用固相萃取-超高效液相色谱-串联质谱的方法对保健食品中的大豆异黄酮进行检测,采用固相萃取技术,去除了油性基质干扰;采用超高效液相色谱分离技术,增加分析的通量,缩短了分析时间,同时减少溶剂用量,降低分析成本[12];通过质谱检测器检测,实现高灵敏度,检出浓度达到纳克数量级。研究结果表明,此种方法样品前处理简单、有机试剂消耗少、时间短,能够满足大豆异黄酮定性和定量检测需求,为保健食品的质量评价及研究提供有价值的参考。
1 材料与方法
1.1 材料与仪器
大豆磷脂软胶囊 汤臣倍健股份有限公司,批号20180401G;倍仕好牌大豆异黄酮软胶囊 浙江康恩贝健康科技有限公司 批号R1810021;百合康牌大豆卵磷脂软胶囊 威海百合生物技术股份有限公司 批号412688012;大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素(纯度均≥90%) 上海安谱实验科技有限公司;甲醇、乙腈(色谱级) 德国Merker公司;甲酸(色谱级) 德国CNW公司;CNWBOND Si、CNWBOND Florisil、CNWBOND HC-C18、CNWBOND LC-C18、Poly-Sery MCX固相萃取柱 上海安谱实验科技有限公司;实验用水由Milli-Q纯水机制得。
XS205DU电子天平 瑞士METTLER TOLEDO公司;API 4000+三重四级杆串联质谱仪 美国AB公司;H-CLASS超高效液相色谱仪 美国Waters公司;2695/2998高效液相色谱仪 美国Waters公司;KH-100B型超声波清洗器 昆山禾创超声仪器有限公司;X1R型高速低温离心机 美国ThermoFisher公司;Milli-Q超纯水机 美国Millipore公司。
1.2 实验方法
1.2.1 样品制备 取20粒软胶囊样品,倾出内容物,用干净玻璃棒搅拌均匀,现用现制。
1.2.2 标准储备溶液的配制 分别称取大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素标准品0.0100 g于100 mL容量瓶中,以甲醇定容至刻度,得100 μg/mL大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素单标储备溶液,4 ℃避光保存。
1.2.3 混合标准使用溶液的配制 分别移取1 mL大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素单标储备溶液于同一100 mL容量瓶中,用甲醇定容得浓度为1 μg/mL混合标准中间溶液。分别移取0.01、0.02、0.05、0.1、0.2、0.5、1、2、5 mL混合标准中间溶液于10 mL容量瓶中,用甲醇定容至刻度,得浓度为1、2、5、10、20、50、100、200、500 μg/L的混合标准使用溶液,以此绘制标准曲线,外标法定量。
1.2.4 样品处理 称取0.5 g样品置于50 mL容量瓶中,加80%甲醇溶液至接近刻度,超声提取20 min,用80%甲醇溶液定容,摇匀。取样品溶液置于离心管中,8000 r/min离心10 min。取上清液1 mL全部通过CNWBOND Florisil固相萃取柱(预先以5 mL甲醇,5 mL水活化),以5 mL水淋洗柱体,25 MPa加压抽干5 min。以6 mL甲醇洗脱柱体,于40 ℃氮气吹干,以1 mL 80%甲醇溶液溶解,过0.22 μm微孔滤膜后待测定。
1.2.5 高效液相色谱串联质谱条件 色谱柱:Waters ACQUITY UPLC C18柱(2.1 mm×100 mm,1.7 μm);流动相:A相为0.1%甲酸水溶液,B相为乙腈;梯度洗脱程序为:88% A 0~4.0 min;88%~80% A 4.0~6.0 min;80% A 6.0~8.0 min;80%~70% A 8.0~10.0 min;70%~20% A 10.0~10.1 min;20% A 10.1~12 min,流速0.3 mL/min,柱温30 ℃,进样体积2 μL。质谱离子源为ESI(+),雾化电压:5500 V;离子传输温度:500 ℃,各大豆异黄酮多反应监测(MRM)模式监测参数,见表1。
1.2.6 定性、定量检测
1.2.6.1 定性检测 取1.2.3 中50 μg/L的混合标准使用溶液和1.2.4中样品溶液按色谱条件1.2.5进行测定,依据标准品色谱峰保留时间,对样品溶液中的组分进行定性,保留时间应一致。
1.2.6.2 定量检测 将1.2.3中大豆异黄酮混合标准使用溶液在色谱条件1.2.5下进行测定,绘制以峰面积为纵坐标、浓度为横坐标的标准曲线。将1.2.4制备的样品溶液注入液质联用仪中,保证样品溶液中大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素的响应值均在工作曲线的线性范围内,若超出最高浓度,将样品稀释后再进样。由标准曲线得到样品溶液中大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素的浓度。
2 结果与分析
2.1 固相萃取柱的选择
样品未经过除油处理,会残留较多油溶性杂质,如亚油酸、油酸等,特别是批量进样后,色谱柱受污染更重,致使系统压力明显升高,很容易超过警戒最高压力[13]。
通过固相萃取能够去除大豆苷之前的大部分杂峰,很好地降低了样液对液相色谱柱的污染损耗,延长了色谱柱的使用时间[14]。实验考察了5种不同填料的固相萃取柱,测定样品在添加1 mL 50 μg/L大豆异黄酮混合标准使用溶液的回收率。表2结果表明Florisil固相萃取柱有较好的回收率和净化效果,大豆异黄酮中各组分回收率最高,平均回收率为85.4%,并具有污染小、可处理小体积试样等优点,能够在保留目标化合物的同时,除去油性样品中的干扰物,可用于大豆异黄酮分离纯化。Florisil 固相萃取柱中的萃取介质弗罗里硅土作为氧化镁复合的极佳硅胶吸附剂,适合于从非极性基质中吸附极性化合物,具有高吸附容量、高灵敏度、方便耐用和稳定等优点[15]。

表2 不同固相萃取柱(SPE)的回收率实验结果Table 2 Experimental results on recovery of different solid phase extraction columns(SPE)
2.2 质谱条件的选择
大豆异黄酮各组分在质谱上具有很强的正离子响应,因此选择正离子模式进行扫描。500 μg/L混合标准使用溶液采用蠕动泵进样分析,在正离子模式下,6种大豆异黄酮各组分一级质谱均出现[M+H]+准分子离子峰,且丰度较大,因此选择分子离子峰作为母离子。分别对6中大豆异黄酮各组分母离子进行轰击碎裂,得到特征碎片离子峰信息,确定各目标物的特征子离子,具体见表1。在MRM模式下优化碰撞能量、去簇电压,确定最佳质谱检测条件。

表1 6种大豆异黄酮质谱分析参数Table 1 MS/MS parameters of 6 soybean isoflavones
2.3 色谱条件的选择
2.3.1 色谱柱的选择 以C18色谱柱作为分析柱。通过系列实验,比较了Waters ACQUITY UPLC C18柱(2.1 mm×100 mm,1.7 μm)、Agilent Eclipse Plus C18(2.1 mm×50 mm,1.8 μm)和Agilent Eclipse XDB-C18(2.1 mm×50 mm,1.8 μm)三种色谱柱对6种大豆异黄酮各组分的分离效果。结果表明,在相同色谱条件下Waters ACQUITY UPLC C18柱对异黄酮的分离效果最好,能提供良好的峰形,因此选择该色谱柱作为分析柱。
2.3.2 流动相的选择 考察了不同流动相对异黄酮色谱行为的影响。以含乙腈-水、甲醇-水作流动相,在采取梯度洗脱的方式时,大豆苷和大豆黄苷没有完全分离开;采用0.5%的乙酸水溶液和甲醇作为流动相,大豆素和大豆黄素没有完全分离开;采用20 mmol/L乙酸铵水溶液和乙腈作为流动相,大豆黄素与染料木素色谱峰重合;以0.1%甲酸水溶液-乙腈为流动相,通过优化两相的配比,6种异黄酮均可获得满意的分离度。通过优化梯度洗脱条件,获得了6个目标物合适的保留时间和峰形,6种大豆异黄酮总离子流图见图1。

图1 6种大豆异黄酮总离子流图Fig.1 Total ion chromatogram of 6 kinds of soybean isoflavones注:1:大豆苷;2:大豆黄苷;3:染料木苷; 4:大豆素;5:大豆黄素;6:染料木素。
2.4 方法的线性关系与检出限
分别将1.2.3的混合标准使用溶液按色谱条件进行测定,以6种大豆异黄酮各组分的色谱峰面积(y)和质量浓度(x)做线性回归,结果显示大豆苷、染料木苷、大豆素以及染料木素在10~500 μg/L范围内,大豆黄苷在5~500 μg/L范围内、大豆黄素在1~200 μg/L范围内线性关系均良好(r>0.99),见表3。以3倍的信噪比确定方法的检出限,大豆黄苷、大豆黄素检出限均为10 μg/kg,定量限均为30 μg/kg,大豆苷、染料木苷检出限均为20 μg/kg,定量限均为60 μg/kg,大豆素、染料木素检出限均为 30 μg/kg,定量限均为90 μg/kg。

表3 6种大豆异黄酮各组分的线性关系与检出限Table 3 Linear relationship and detection limit for 6 soybean isoflavones
2.5 方法的回收率和精密度考察
在上述优化后的试验条件下,分别对样品添加2、5和10 mg/kg的标准品后进行测定,平均回收率和精密度见表4。结果显示,加标回收率为81.8%~98.4%,相对标准偏差为1.8%~6.7%,能够满足检测方法的要求。

表4 样品中6种大豆异黄酮各组分的加标回收率和精密度(n=6)Table 4 Results of recovery and repeatability for 6 soybean isoflavones(n=6)
2.6 实际样品中6种大豆异黄酮各组分检测
取汤臣倍健大豆磷脂软胶囊、倍仕好牌大豆异黄酮软胶囊、百合康牌大豆卵磷脂软胶囊,按照标准GB/T 23788-2009[11]和本文方法进行检测,样品中大豆异黄酮含量的测定结果见表5。结果表明,标准GB/T 23788-2009方法检出限为5 mg/kg,样品中的大豆苷、大豆黄素若含量低于5 mg/kg,则未检出,而且在进样过程中,系统压力过大,两次报错,说明前处理方法不合理,未经去油处理直接进样会堵塞色谱柱,而超高效液相色谱串联质谱检出限为10~30 μg/kg,低含量的物质也可以精确定量,通过Florisil固相萃取柱净化,去除了油性基质干扰,整个进样过程中,系统压力稳定。两种方法的检测结果偏差在2.2%~8.3%,结果可信。
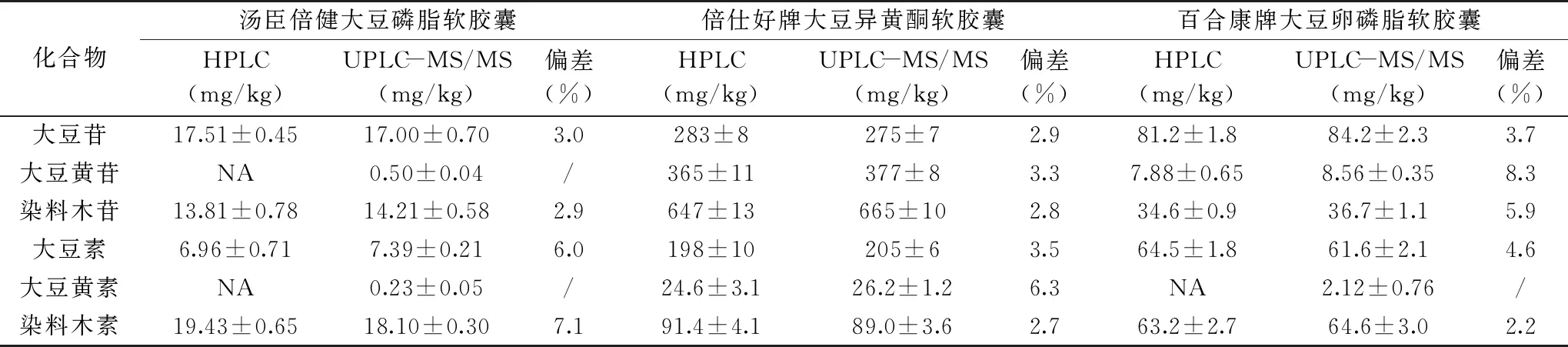
表5 样品中大豆异黄酮含量的测定结果Table 5 Determination results of soybean isoflavones contents in the samples
3 结论
建立了保健食品中大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素超高效液相色谱串联质谱同时检测方法。结果表明,大豆苷、大豆黄苷、染料木苷、大豆素、大豆黄素以及染料木素在各自浓度范围内线性关系良好;大豆黄苷、大豆黄素检出限均为10 μg/kg;大豆苷、染料木苷检出限均为20 μg/kg;大豆素、染料木素检出限均为30 μg/kg,加标回收率为81.8%~98.4%,相对标准偏差为1.8%~6.7%。通过与标准GB/T 23788-2009液相方法结果比较,该方法具有较高的灵敏度和较低的检出限,在实际检测工作中具有良好应用价值。
猜你喜欢
海峡姐妹(2019年8期)2019-09-03
天津科技大学学报(2019年2期)2019-04-22
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年5期)2018-06-13
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年6期)2017-06-13
中国造纸(2017年2期)2017-04-07
中医研究(2014年4期)2014-03-11
中国中医药现代远程教育(2014年21期)2014-03-01
