
VH3分子稳定构型的理论研究
2019-09-10 20:22任桂明
读与写·教师版 2019年2期
任桂明

摘要:本文用密度泛函中的B3LYP和BP86方法在DZP全电子基组水平上对VH3分子的较低能量构型及其对称性、电子态、总能量、相对能量、电偶极矩、键长、键角等进行了理论计算,结果表明:两种方法计算所得VH3分子都具有三个较低能量构型,基态构型结构几乎一致,均属D3h对称点群,其余构型具有C2v对称性。
关键词:密度泛函(DFF);钒氢化合物;稳定构型
中图分类号:0641.1 文献标识码:A 文章编号:1672-1578(2019)02-0292-01
1.引言
过渡金属钒在电子能谱学、胶体稳定性、电极材料等方面倍受青睐。对过渡金属钒的理论研究上,Salahub和Messmer用自旋极化自洽场(sCF)-xa-SW方法计算了钒团簇的性质。秦渝,方志刚等人采用密度泛函理论,研究了团簇V,P2的稳定结构和电子性质。贾美叶,何圣贵等人对氧化钒团簇正离子与硫化氢反应的实验和理论研究。Su等人通过实验得到了钒团簇的键离解能。研究钒的氢化物的结构及其形成机理目前鲜见文献报道。本文的目的是利用密度泛函中的B3LYP和BP86的方法对VH3的稳定结构的总能量、对称性、电子态等进行理论研究和计算,为进一步研究钒氢化物体系提供理论参考。
2.计算方法
先将一个钒原子、三个氢原子组成各种具有高对称性的不同的空间结构,再分别调整钒氢原子之间的键长、键角和二面角等参数,在Windows环境下,分别用B3LYP和BP86方法在DZP全电子基组水平上对VH3分子进行理论研究和计算。
3.结果与讨论
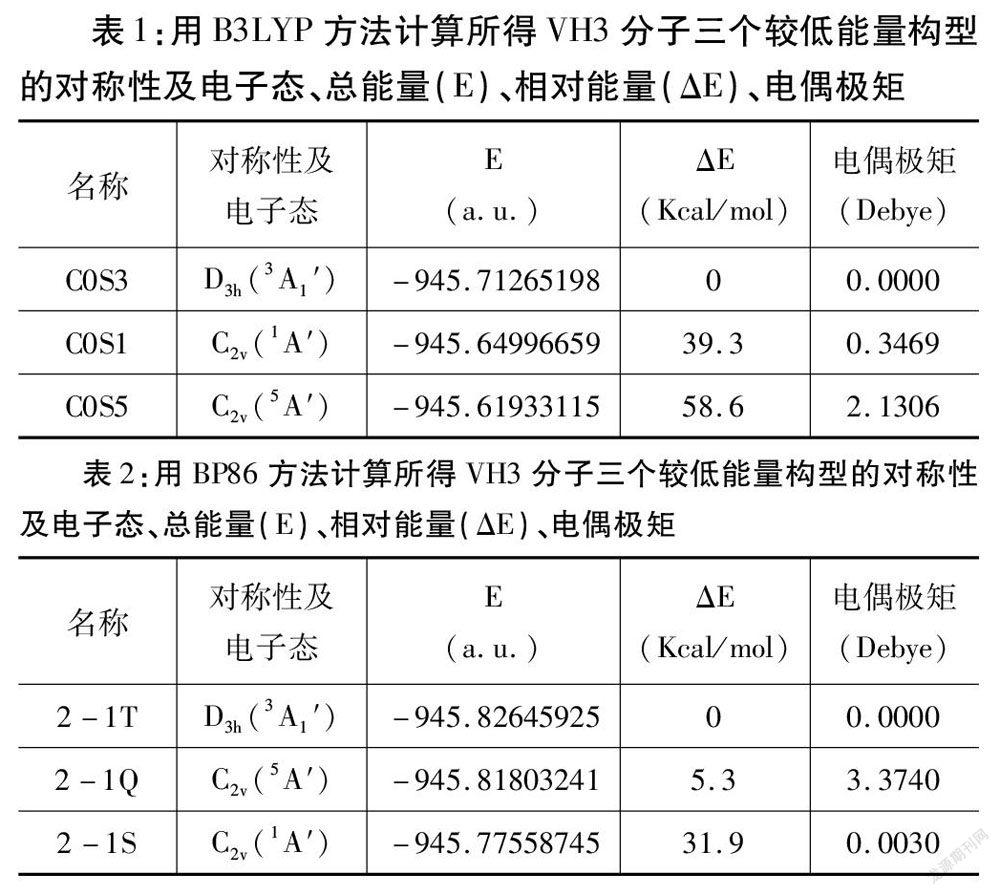
图1给出了用两种密度泛函方法计算得到的VH3分子的基态构型(即COS3和2-1T),基态构型V-H之间的键长都为1.675A,键角为120°,它是一个具有较高对称性的结构。表1和表2分别给出了用B3LYP和BP86方法计算得到的VH3分子三个较低能量构型的对称性及电子态、总能量(E)、相对能量(AE)、电偶极矩。表1中COS3结构能量为:-945.71265198a.u.,具有Dsh对称性,电偶极矩为:ODebye,该分子为非极性分子。从能量的角度看:该结构的能量是最低的,为基态结构。比基态构型能量高39.3Kca/mol的一重态构型COSl,具有C2v对称性,电偶极矩为:0.3469Debye,该分子为弱极性分子。COS5构型,能量比基态能量高58.6Kcal/mol,具有C2v对称性,电偶极矩为:2.1306Debye,为极性分子。表2中2-1T结构能量为:一945.82645925a.u.,具有D3h对称性,电偶极矩为:0Debye,该分子为非极性分子。从能量的角度看:该结构的能量是最低的,该结构为基态结构。比基态的能量高5.3Kcal/mol的2-1Q構型,具有C2v对称性,它的电偶极矩为:3.3740Debye,该分子为极性分子,图1可知该构型还有两个特殊的H原子,不但与金属V原子连接,两个H原子还相互连接,含有一个氢分子。一重态的2—1s构型的能量比基态高31.9 Kcal/mol,具有c2,对称性,电偶极矩为:0.0030 Debye,为弱极性分子。
4.结语
本文运用两种密度泛函方法,从理论上预测了VH3分子的较低能量构型,计算出的所有结构均无虚频。从能量的角度分析了VH3分子的稳定性,VH3分子的基态为三重态,即本文中的COS3、2-1T结构。用B3LYP方法计算所得的一重态构型COSI和五重态构型COS5,其能量都比基态分子能量高出30Kcal/mol以上,而用BP86方法计算所得的五重态构型2—1Q能量仅比基态构型高5.3Kcal/mol,而一重态的2-1S构型能量比基态能量高31.9Kcal/mol。基态构型对称性为D3h,电子态为:A1’,都为非极性分子,且V与H原子之间的键长都在单键长度要求的范围内。
