
层状K-Fe-Zn-Ti催化剂的制备及其对二氧化碳加氢制烯烃反应的催化性能
2019-09-04 07:24:30吴大凯高新华马清祥张建利范素兵赵天生
燃料化学学报 2019年8期
吴大凯, 王 旭, 高新华, 马清祥, 张建利, 范素兵, 赵天生
(宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室, 宁夏 银川 750021)
CO2化学转化制高附加值化学品是实现其资源化利用的一条重要途径[1]。近年来,CO2加氢直接制烯烃成为研究热点之一。该过程可由逆水煤气变换(Reverse Water Gas Shift, RWGS)反应(CO2+H2→ CO+ H2O),再经费托合成反应(Fischer-Tropsch Synthesis, FTS)发生C-C偶联(CO+H2→(-CH2-)+H2O)路线实现,简称CO2-FTS过程[2,3]。Fe基催化剂在CO2加氢反应中具有高活性和烯烃选择性,是目前经费托路线研究的主要催化体系,Fe3O4和FeCx分别为RWGS反应和FTS反应的活性相[4,5]。如何实现两步反应的协同以获得高烯烃选择性是技术关键。FTS反应初级产物富含烯烃,极易发生加氢、异构化等二次反应,导致烯烃选择性下降。抑制初级烯烃的二次反应,可达到调控产物分布,提高烯烃选择性的目的[6,7]。因此,设计一种双功能催化剂在实现两步反应协同的同时,有效抑制烯烃的二次反应,有望进一步提高烯烃选择性。
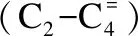
基于前期研究,本研究采用高温固相反应,制备了Zn改性的四组元层状K-Fe-Zn-Ti催化剂,用于CO2加氢制烯烃反应,结合对催化剂的系统表征,探究了Zn的添加对CO2加氢产物分布、特别是烯烃选择性的影响规律和作用机理。
1 实验部分
1.1 催化剂的制备
以K2CO3、Zn(NO3)2·6H2O、Fe(NO3)3·9H2O、TiO2为原料,按一定配比分别称取一定质量的药品,充分混合后在500 ℃下焙烧3 h;所得粉末经研磨后,在管式炉中采用下述升温程序:25-500 ℃,5 ℃/min;500-800 ℃,10 ℃/min;800-1000 ℃,5 ℃/min;1000-1100 ℃,2 ℃/min,再经1100 ℃焙烧10 h,自然冷却至室温。焙烧后样品经充分研磨后,压片造粒至20-40目备用。新鲜样品记为K-Fe-Zn-Ti,反应后记为AR-K-Fe-Zn-Ti。
1.2 催化剂的表征
催化剂物相表征采用德国布鲁克AXS有限公司X射线衍射仪(XRD),型号D8 ADVANCE A25,CuKα辐射源,λ= 0.154 nm的光源波长,管电压40 kV,管电流40 mA,3°-85°扫描、扫描速率8(°)/min,步长0.02°。
催化剂的N2物理吸附在美国康塔仪器公司上进行测试。测试前样品在真空度0.1 Pa以下300 ℃处理2 h清除催化剂表面杂质,在液氮温度77 K、N2分压10-8-10-1进行等温吸附实验。
在美国TA仪器公司SDTQ600上对催化剂进行热重分析,将8 mg样品置于耐热坩埚内在空气气氛(流量为30 mL/min)下以10 ℃/min的速率升温至1100 ℃。
催化剂的形貌表征在ZEISS EVO18型钨灯丝扫描电子显微镜(SEM)上进行,测试前对催化剂进行喷金处理,工作电压为3.0 kV。透射电镜(TEM)表征在Tecnai G2 F30上进行。
H2-TPR测试采用天津先权公司TRP-6060多用吸附仪。将0.05 g催化剂置于石英管中,He气氛下350 ℃吹扫40 min,降至室温,切换至5% H2-N2气氛,以10 ℃/min的升温速率升至800 ℃,同步记录还原曲线。
CO2-TPD使用美国麦克公司AutoChem Ⅱ2920型多用化学吸附仪。高纯He为载气,流量30 mL/min,催化剂装填量0.05 g;30% H2(体积分数)还原,从室温以10 ℃/min速率升至400 ℃,还原0.5 h,降至室温,吹扫1 h;升温至50 ℃,脉冲吸附CO2至饱和,继续吹扫1 h,之后10 ℃/min升温至800 ℃,记录脱附CO2-TPD谱图。
使用赛默飞世尔科技公司的ESCALAB 250型光谱仪进行XPS测试,射线源AlKαX,分析室真空度为2×10-7Pa,以C 1s284.8 eV为准进行峰位校正,根据Cu 2p3/2,3p峰对仪器校正,管电压为15 kV,管电流为10 mA。
1.3 催化剂的性能评价
CO2加氢性能测试采用内径为8 mm的小型固定床反应器。催化剂装填量1 mL。在线还原条件:H2、1000 h-1、0.1 MPa、400 ℃还原6 h;反应条件:H2/CO2(体积比) = 3、320 ℃、2 MPa、1000 h-1。气相产物经GC-9160-I色谱仪在线分析,分别采用长2 m的TDX-01柱和长50 m的Al2O3毛细管柱分析C1和C1-5烃组分;液相产物累计取样后经GC-9160-II色谱仪离线分析,分别采用长2 m的TDX-401柱和长30 m毛细管SE-30柱分析水相和油相产物。气相产物采用甲烷关联法定量,液相产物采用面积归一法定量。
2 结果与讨论
2.1 催化剂的物相
图1为反应前催化剂样品的XRD谱图。
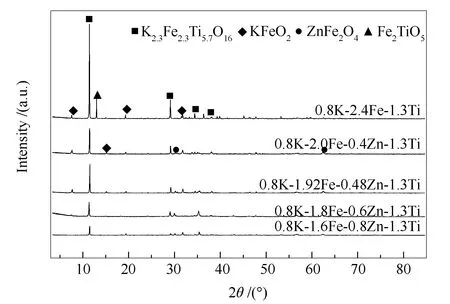
图 1 反应前催化剂样品的XRD谱图
由图1可知,在所考察的配比范围内,经1100 ℃焙烧后,样品均具有典型的LMO结构[18],在11.36°、29.18°、34.3°、38.16°出现LMO的特征衍射峰,为K2.3Fe2.3Ti5.7O16物相[19]。在7.4°、14.9°、19.1°、31.5°为KFeO2;在12.5°出现Fe2TiO5物相。0.8 K-2.4Fe-1.3Ti样品的LMO结构特征衍射峰较强且尖锐,Zn改性后,在30°、35.3°、42.5°处出现ZnFe2O4物相[20],LMO结构衍射强度下降,且随Zn含量的增加,趋势越明显。
图2为反应后催化剂样品的XRD谱图。由图2可知,反应后催化剂仍保持LMO结构,具有良好的结构稳定性;与反应前相比,ZnFe2O4衍射峰强度略有增加,且Zn改性的催化剂均在43°左右出现较弱的Fe3C衍射峰[21],表明反应过程中铁物相发生了部分碳化。
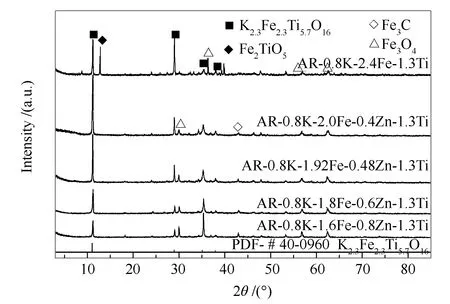
图 2 反应后催化剂样品的XRD谱图
2.2 催化剂的形貌
图3为反应前催化剂样品的SEM照片。

图 3 新鲜样品的SEM照片
由图3可知,所有样品的层状形貌明显,均由片状和块状堆积而成[22],长度约1 μm,厚度0.10-0.20 μm。0.8K-2.4Fe-1.3Ti样品(图3(a))结晶度较好,片层结构明显;Zn改性后催化剂形貌发生一定程度的改变,随Zn含量的增加,样品结晶度下降,出现不均匀的球型颗粒,结合XRD(图1)分析,可能与新生成的ZnFe2O4物相有关。当Zn/Fe=1∶2时,样品以块状堆积在一起,大小不均匀,结晶度差。
图4为反应前后0.8K-1.92Fe-0.48Zn-1.3Ti样品的TEM照片。由图4可知,反应前后样品的层状结构得以保持,为K2.3Fe2.3Ti5.7O16的(020)晶面(PDF-#JCPDS 40-0960),反应前后层间距不变,为0.78 nm。

图 4 0.8K-1.92Fe-0.48Zn-1.3Ti催化剂的TEM照片Figure 4 TEM images of 0.8K-1.92Fe-0.48Zn-1.3Ti catalyst(a): fresh sample; (b): used sample
2.3 催化剂的织构性质
图5、表1分别为催化剂样品的N2吸附-脱附曲线,比表面积、孔径和孔体积数据。由图5可知,所有样品吸附等温线均为IV型等温线,在p/p0为0.3-1.0出现回滞环,表明所有样品均具有介孔结构。由表1可知,所有样品均具有较小的比表面积;随Zn含量的增加,催化剂的比表面积呈先增大后减小后变化趋势;相对较大的比表面积有利于在反应中暴露更多活性位点,促进反应传质、提高催化活性[23];Zn改性后孔体积有所降低,平均孔径增大。
图 5 催化剂的N2吸附-脱附等温曲线

表 1 催化剂的织构性质
2.4 热重分析
图6为反应后催化剂样品AR-0.8K-1.92Fe-0.48Zn-1.3Ti的热重谱图。由图6可知,样品失重过程可分为三个阶段:第一阶段在50-150 ℃,质量损失约为0.82%,失重对应于表面、层间吸附水的挥发;在240-570 ℃的第二阶段,质量损失约占4.43%,可能是由于催化剂表面吸附的低碳数烃的分解,对应的DSC曲线有一个明显的放热峰;570 ℃以上为第三阶段,570 ℃以上失重可能来自于催化剂表面吸附的高碳数烃或积炭的分解。
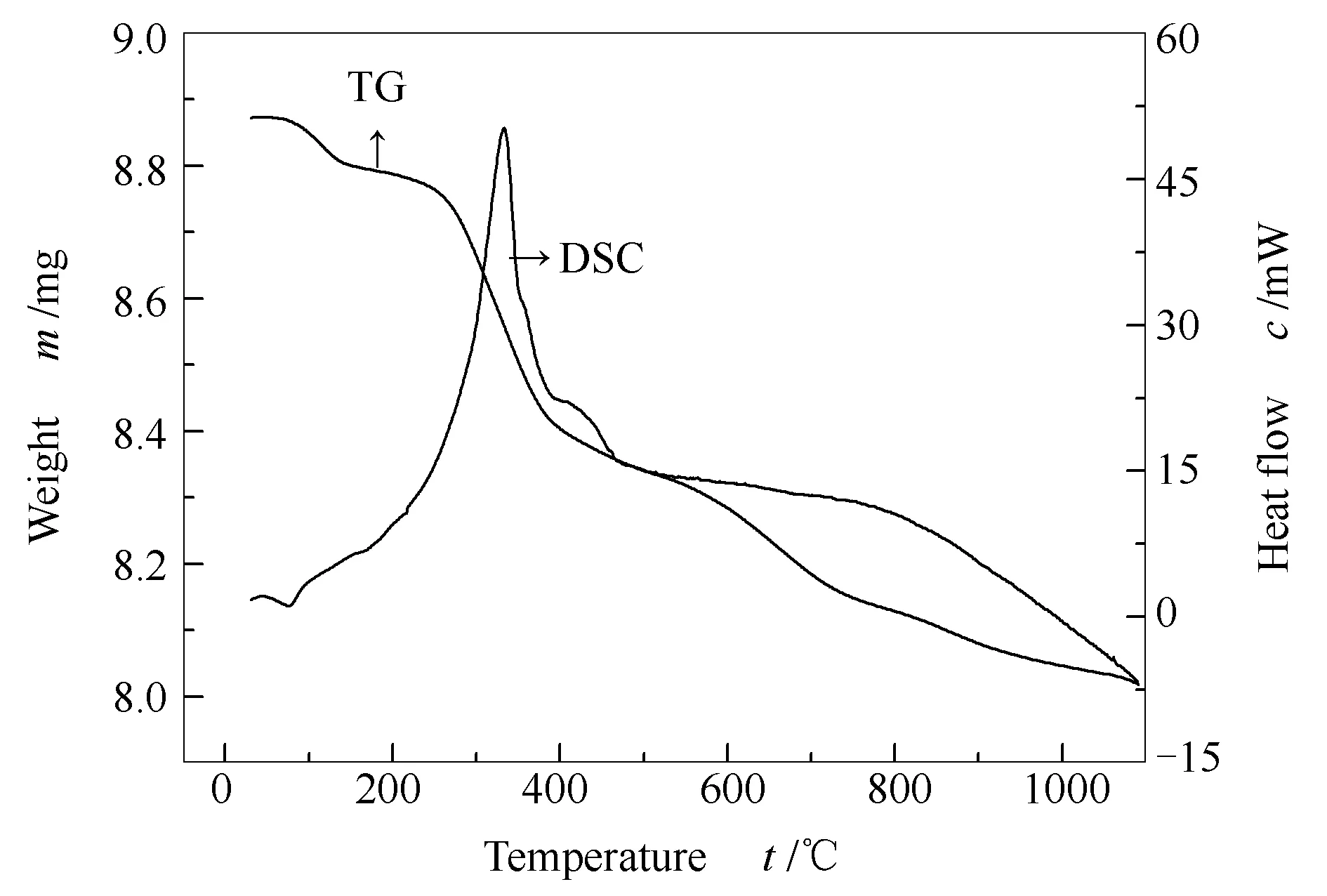
图 6 AR-0.8K-1.92Fe-0.48Zn-1.3Ti的热重分析曲线
2.5 还原性能
图7为催化剂样品的H2-TPR谱图。由图7可知,Zn改性催化剂样品出现两个耗氢峰;低温(400-580 ℃)耗氢峰归属为Fe2O3→Fe3O4的还原,高温(600-850 ℃)耗氢峰归属为Fe3O4→FeO→α-Fe的还原[24]。0.8K-2.4Fe-1.3Ti样品未见明显的Fe2O3→Fe3O4还原峰;随Zn含量的增加,Fe2O3→Fe3O4还原峰向低温方向偏移,表明助剂Zn促进了样品还原,有利于Fe碳化物的生成,进而促进FTS成烃反应的进行。但与Fe2O3的还原相比[25],K-Fe-(Zn)-Ti LMO结构催化剂中Fe的还原明显受到抑制。
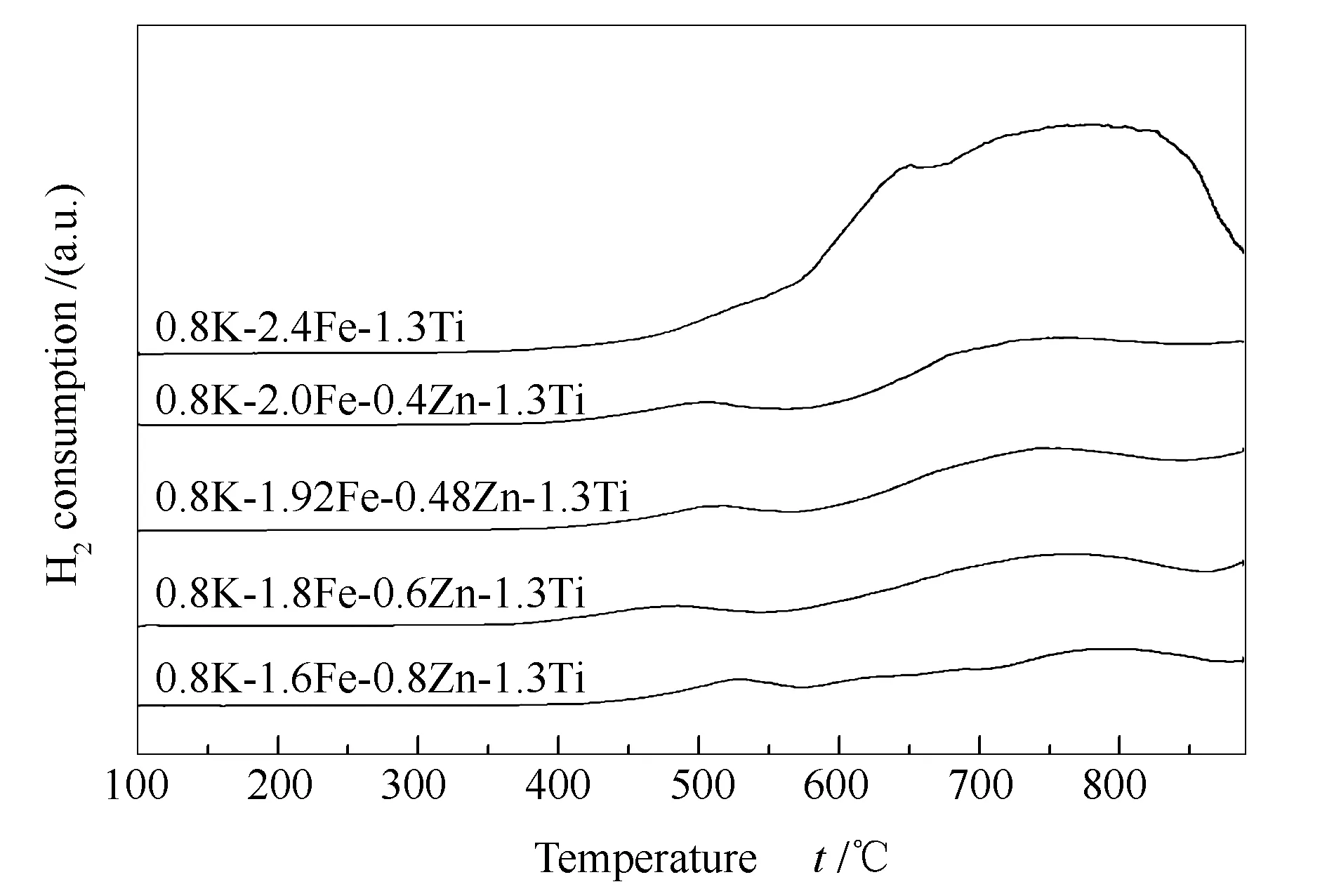
图 7 催化剂样品的H2-TPR谱图
2.6 催化剂表面碱性
图8为催化剂样品的CO2-TPD谱图。由图8可知,所有样品均出现两个脱附峰,即在100 ℃左右的弱吸附和600-700 ℃的强吸附,对应两种不同CO2吸附强度的吸附位[26]。102 ℃附近的低温脱附峰对应于CO2的物理脱附,650 ℃左右的脱附峰为CO2的化学吸附,对应于催化剂样品的表面强碱性位点[2,27]。Zn改性后650 ℃左右脱附峰峰面积增加,且均向高温方向移动,表明Zn改性提高了催化剂表面碱性,增强了CO2表面吸附。
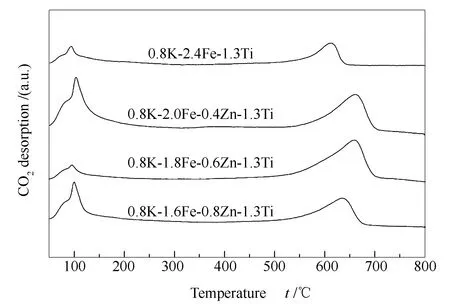
图 8 催化剂样品的CO2-TPD谱图
2.7 XPS表征
图9分别为几种不同Zn/Fe比催化剂的Fe 2p、Ti 2p的XPS谱图。
由图9可知,Zn助剂的供电子效应导致随Zn含量的增加,Fe、Ti电子结合能均向低结合能方向偏移。Fe 2p谱图(图9(a))表明,新鲜催化剂均在711.12、724.26 eV处出现Fe2O3的Fe 2p3/2和2p1/2特征峰[28-30],与新鲜样品相比,反应后Fe 2p结合能进一步降低至710.9 eV,结合XRD(图2)分析,证明有Fe3O4生成。新鲜样品的Ti 2p谱图(图9(b))与Fe 2p变化规律相似,但反应后Ti 2p结合能位置向高温方向移动,可能与较为稳定的ZnFe2O4物相生成有关,减弱了Zn与Ti相互作用。反应后,催化剂表面大量积炭(见表1),导致Fe 2p、Ti 2p谱图信号降低[10,22]。
表2为不同配比的催化剂样品反应前后表面含量组成的变化。
由表2可知,催化剂经Zn改性后,表面K元素含量与未改性催化剂相比有所降低。随Zn/Fe比增加,催化剂表面Fe含量逐渐降低,但Ti含量基本保持不变。反应后,样品表面Fe、Ti含量明显下降,表面C含量显著增加,表明反应后催化剂表面大量积炭,且出现表面K富集。
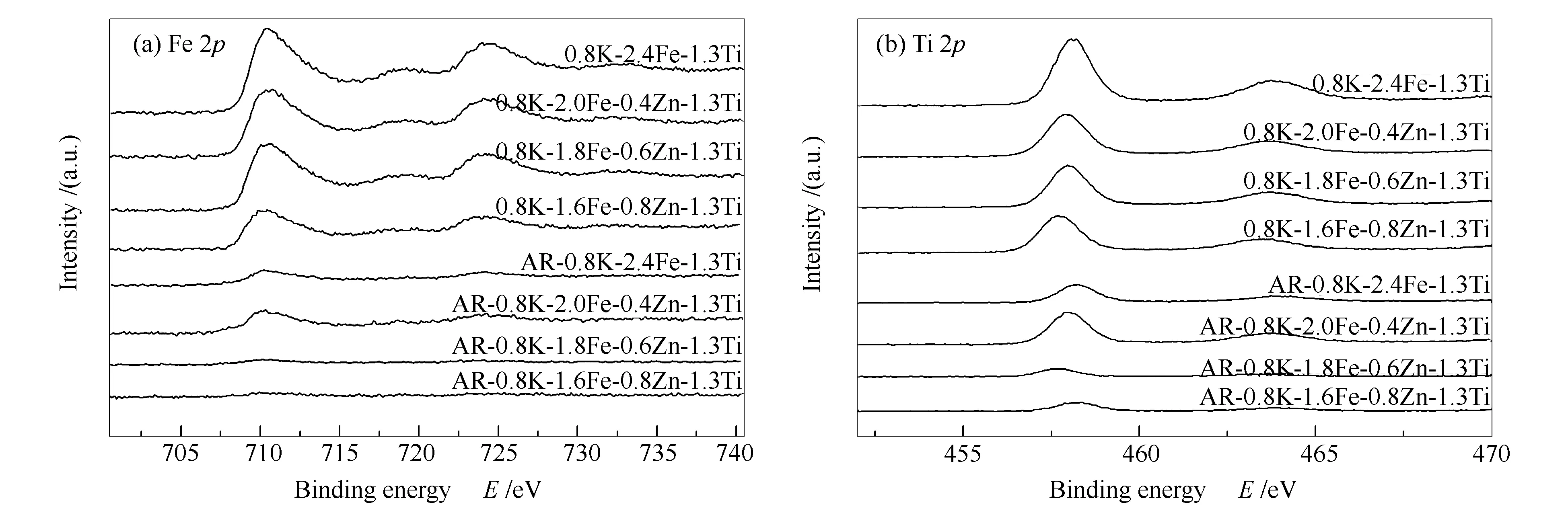
图 9 反应前后催化剂样品的XPS谱图Figure 9 XPS spectra of the K-Fe-Zn-Ti catalysts
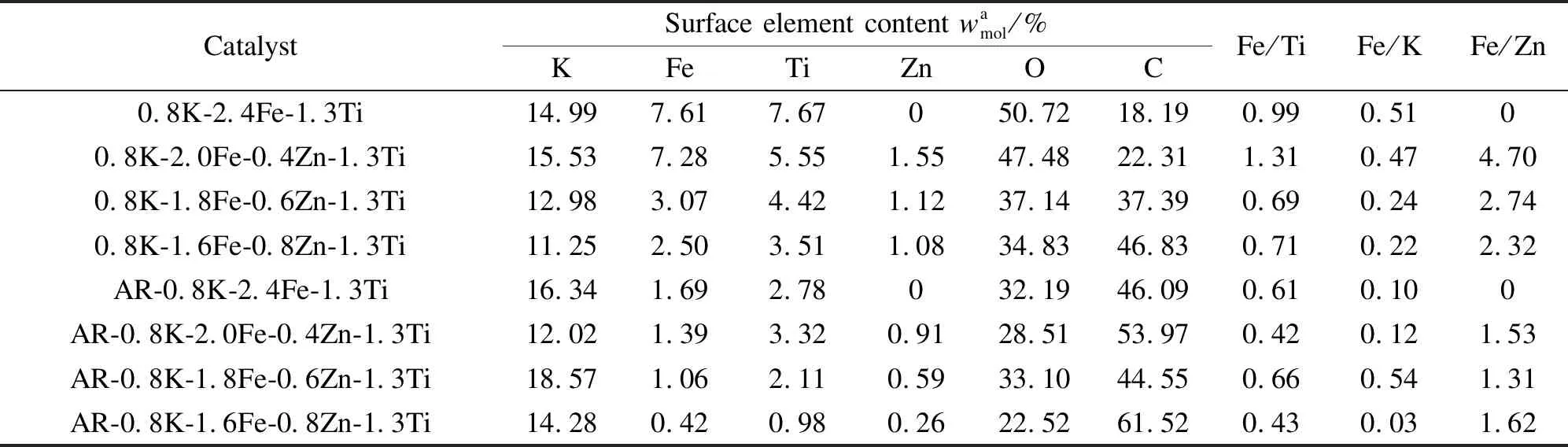
表 2 反应前后催化剂样品的表面组成
a: calculated from peak area of XPS spectra
2.8 催化活性评价
表3为不同配比催化剂的CO2加氢活性评价数据。
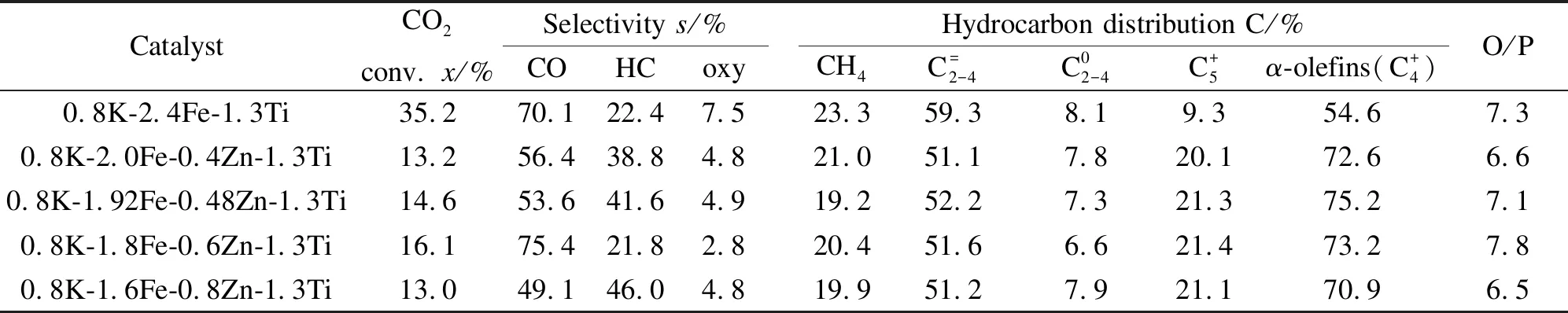
表 3 催化剂的活性评价Table 3 Performances of the K-Fe-Zn-Ti catalysts in CO2 hydrogenation
reaction condition: 320 ℃, 2 MPa, H2/CO2= 3.0, GHSV = 1000 h-1, TOS = 72 h

3 结 论
猜你喜欢
云南化工(2021年8期)2021-12-21 06:37:38
中国石油石化(2021年9期)2021-07-17 09:24:10
世界有色金属(2020年4期)2020-05-16 05:55:44
中国塑料(2016年12期)2016-06-15 20:30:07
当代化工研究(2016年2期)2016-03-20 16:21:18
上海金属(2015年6期)2015-11-29 01:08:49
中国塑料(2015年11期)2015-10-14 01:14:14
中国塑料(2015年9期)2015-10-14 01:12:17
中国塑料(2015年4期)2015-10-14 01:09:19
石油炼制与化工(2014年10期)2014-09-16 03:52:04
