
蒙药饮片煨诃子的质量标准研究
2019-09-02 08:04:12敖格日乐图孙丽君肖志彬王玉华那生桑
安徽医药 2019年9期
敖格日乐图,孙丽君,肖志彬,王玉华,那生桑
作者单位:1内蒙古自治区国际蒙医医院药学部,内蒙古自治区 呼和浩特 010065;2内蒙古医科大学药学院,内蒙古自治区 呼和浩特 010110
诃子为使君子科植物诃子(Terminalia chebula Retz.)或绒毛诃子(Terminalia chebula Retz.var.tomentella Kurt.)的干燥成熟果实,分布在我国云南、广西等地[1],主要含三萜类、鞣质类、番泻苷A、诃子素等化学成分,具有涩肠止泻、敛肺止咳、降火利咽的功效。据《中国药典》(2015版)及各地方炮制规范(内蒙古、四川、广西、甘肃等)[2-5],诃子主要的炮制方法有净制、切制、煨制、炒制、蒸制、砂烫制、炒炭等。其中,煨制后鞣质含量显著增高,收涩作用增强[6]。前期实验中,在传统工艺的基础上,通过正交试验已优选出诃子面煨制的工艺条件[7-8],得到不同批次的煨诃子。因此,本实验自2017年5月至2018年5月对煨诃子质量标准进行系统研究,为蒙药材诃子煨制工艺的规范化提供了理论依据。
1 仪器与试药
1.1 仪器硅胶G薄层板(青岛海洋化工有限公司),DV215CD型十万分之一电子分析天平(奥豪斯仪器上海有限公司),Dionex Ultimate3000型高效液相色谱仪(赛默飞世尔科技有限公司),KQ-500E型超声波清洗器(昆山市超声仪器有限公司),AR224CZ电子天平(奥豪斯仪器上海有限公司)。
1.2 试药诃子药材按《中国药典》进行检验符合诃子项下的有关规定;没食子酸对照品(中国食品药品检定研究院,批号:110831-201605)、诃子对照药材(中国食品药品检定研究院,批号:121206-201508);甲醇、乙腈、磷酸均为色谱纯,水为重蒸水,其余试剂均为分析纯。
2 方法与结果
2.1 制法在传统工艺的基础上[5],通过正交试验已优选出诃子面煨制的工艺条件,得煨诃子制法。即取诃子,照煨法(本附录Ⅱ[4]),用面皮包裹约2~3 mm厚,晾至半干,在烫砂中埋煨(410±10)℃,7 min,面皮呈焦黄色时取出(每100 kg诃子,用面粉300 kg)。
2.2 性状本品形状如诃子,质地较酥脆。具焦香气,味酸、极涩。
2.3 显微鉴别本品粉末黄褐色,显微特征同诃子。粉末中以纤维束、石细胞、木化厚壁细胞、靴状厚壁细胞以及草酸钙簇晶为主要显微特征。见图1。

图1 煨诃子粉末显微特征(×400):A为纤维束;B为厚壁细胞;C为石细胞;D为为簇晶
2.4 薄层色谱鉴别
2.4.1对照药材的薄层色谱鉴别 取煨诃子粉末,照《中国药典》(2015年版)诃子项下进行薄层色谱鉴别。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。见图2。
2.4.2没食子酸的薄层色谱鉴别 取煨诃子(去核)粉末0.5 g,加50%无水乙醇30 mL,加热回流30 min,滤过,滤液,蒸干,残渣用甲醇3 mL溶解,作为供试品溶液。另取没食子酸对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述两种溶液各4 μL,分别点于同一硅胶G薄层板上,以甲苯-冰醋酸-水(12∶10∶0.4)为展开剂,展开,取出,晾干,喷以1%三氯化铁乙醇溶液,在日光下检视。供试品色谱中,在与对照品相应的位置上,显相同颜色的斑点。结果见图3。

图2 煨诃子薄层鉴别图:1为煨诃子1(批号:150801;产地:广西);2为煨诃子2:(批号:151002313;产地:云南);3为煨诃子3(批号:1508001;产地:广东);4为没食子酸对照品;5为诃子对照药材

图3 煨诃子薄层鉴别图:1为煨诃子1(批号:150801;产地:广西);2为煨诃子2(批号:151002313;产地:云南);3为煨诃子3(批号:1508001;产地:广东);4为没食子酸对照品
2.5 检查[3]
2.5.1水分 分别称取3批次煨诃子粉末约2.0 g,每批2份,照水分测定法(通则0832第二法)进行测定。结果见表1。

表1 煨诃子的水分测定结果
结果表明,3批次供试品的水分为7.03%~7.40%,结合诃子该项下的规定,故拟定水分为13.0%。
2.5.2总灰分 分别称取3批次煨诃子粉末约2.0g,每批2份,照灰分测定法(通则2302)进行测定。结果见表2。
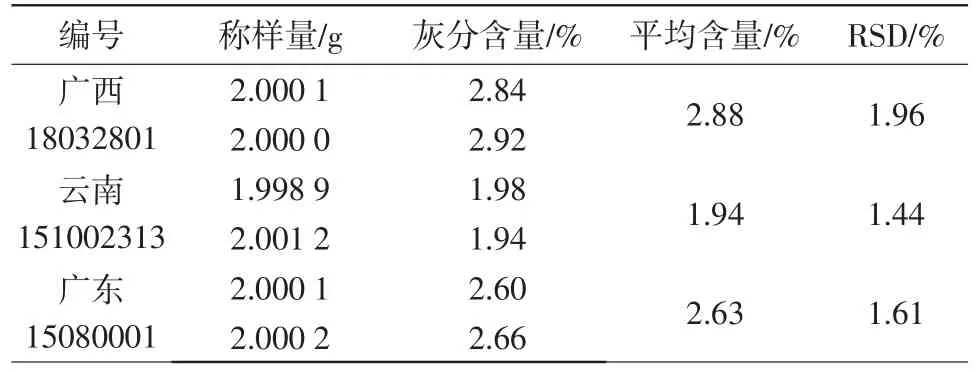
表2 煨诃子的灰分测定结果
结果表明,3批次供试品的灰分为1.94%~2.88%,结合诃子该项下的规定,故拟定灰分为5.0%。
2.5.3浸出物 分别称取3批次煨诃子(批号见表1)粉末约2.0 g,每批2份,照水溶性浸出物测定法(通则2201)项下的冷浸法测定。结果见表3。

表3 煨诃子浸出物测定结果
结果表明,3批次供试品的水溶性浸出物为42.21%~61.34%,结合诃子该项下的规定,故规定水溶性浸出物不得少于30.0%。
2.6 含量测定
2.6.1色谱条件与系统适用性试验 色谱柱为Apollo C18柱(4.6 mm×250 mm,5 μm);以甲醇-0.2%磷酸水溶液(8∶92)为流动相;检测波长为270 nm;流速为1.0 mL/min;柱温25℃。理论塔板数按没食子酸峰计算应不低于3 000。
2.6.2对照品储备溶液的制备 取没食子酸对照品适量,精密称定,加甲醇制成浓度为0.40 mg/mL的溶液,即得。
2.6.3供试品溶液的制备 称取本品粉末(过四号筛)约0.5 g,精密称定,置具塞锥形瓶中,精密加入甲醇溶液25 mL,称定重量,超声处理30 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,过滤,取续滤液,即得。
2.6.4线性关系考察 精密吸取对照品储备液4mL,置10 mL量瓶中,加水稀释至刻度。精密吸取2、5、10、12、15、17、20 –L,分别注入液相色谱仪中,按上述色谱条件测定,记录各图谱的没食子酸峰面积积分值(Y)。以进样量(X:μg)对峰面积积分值(Y)进行回归分析,回归方程为Y=35.107X–0.015 7,r=1。结果见表4和图4。
结果显示,没食子酸在0.32~3.2 μg范围内与峰面积积分值呈良好的线性关系。

表4 标准曲线测定数据(n=7)
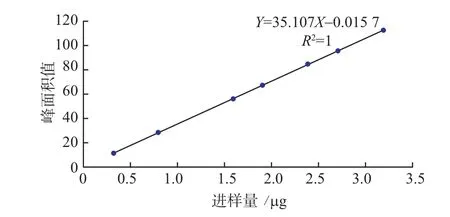
图4 没食子酸标准曲线图
2.6.5精密度试验 取供试品约0.5 g,精密称定,按“2.6.3”项下方法操作,制备供试品溶液,进样5 μL,按上述色谱条件测定,连续进样6针。结果见表5。

表5 精密度试验结果(n=6)
结果显示,RSD=0.28%(n=6),表明该方法精密度良好。
2.6.6重复性试验 取同一批次的供试品粉末约0.5 g,精密称定,共6份。分别按“2.6.3”项下方法操作,制备供试品溶液,进样5 μL,按上述色谱条件测定。结果见表6。
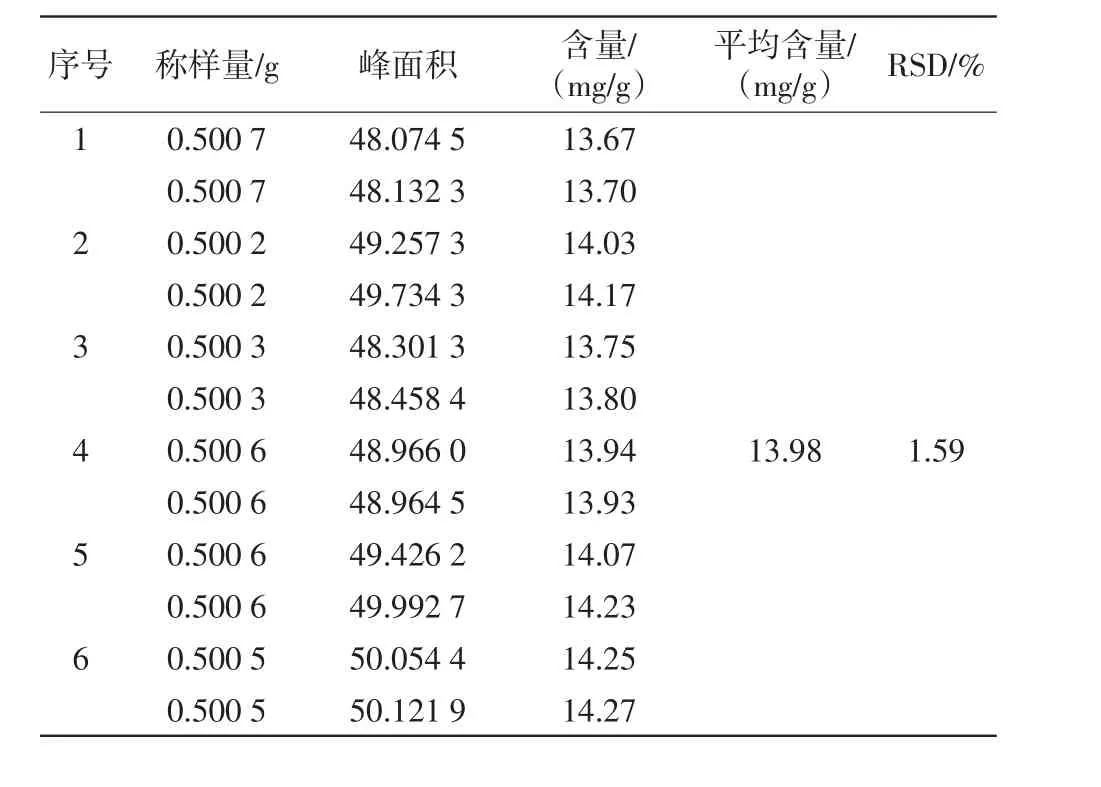
表6 重复性试验结果(n=6)
结果显示,RSD=1.59%(n=6),表明该方法重复性良好。
2.6.7稳定性试验 取同一份供试品溶液,分别在0、1、2、4、6、8、12 h测定。结果见表7。
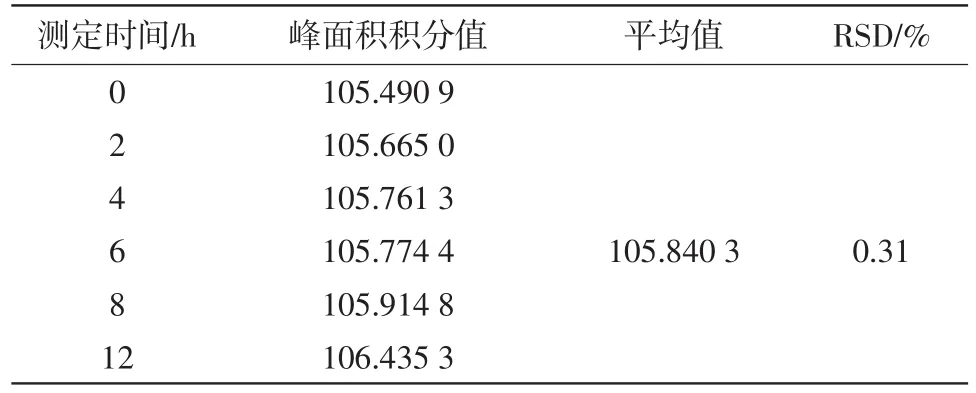
表7 稳定性试验结果
结果显示,RSD=0.31%(n=6),表明供试品溶液在12 h内稳定。
2.6.8加样回收率试验 取已知含量供试品(批号:1508001)约0.25 g,精密称定,共6份。精密加入没食子酸对照品约3.496 mg,照“2.6.3”项下方法操作,测定没食子酸含量,并计算加样回收率。结果见表8。
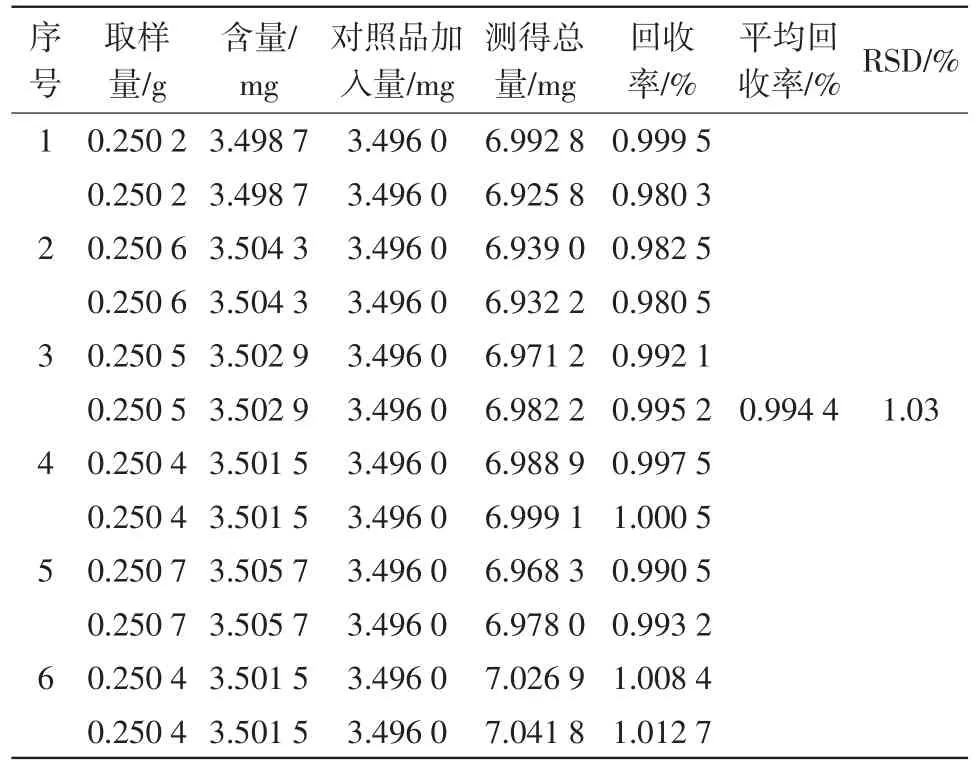
表8 加样回收试验结果(n=6)
结果显示,该方法平均回收试验为99.44%,RSD=1.03%(n=6),表明方法准确度良好。
2.6.9样品含量测定 分别称取上述3批次煨诃子粉末约0.5 g,精密称定,按“供试品溶液的制备”方法操作,制备供试品溶液,进样5 μL,按上述色谱条件测定。结果见表9。
3 讨论
3.1 薄层色谱鉴别煨诃子炮制后药性缓和,涩敛之性增强,主要源于鞣质类成分、番泻苷A等化学成分的变化,对三萜酸类成分影响较小,故本文以生品诃子对照药材为对照对其进行TLC鉴别,此为方法一。但该法没有检出没食子酸,而没食子酸为煨诃子主要有效成分,故本文又以没食子酸对照品为对照对其进行TLC鉴别,即方法二。
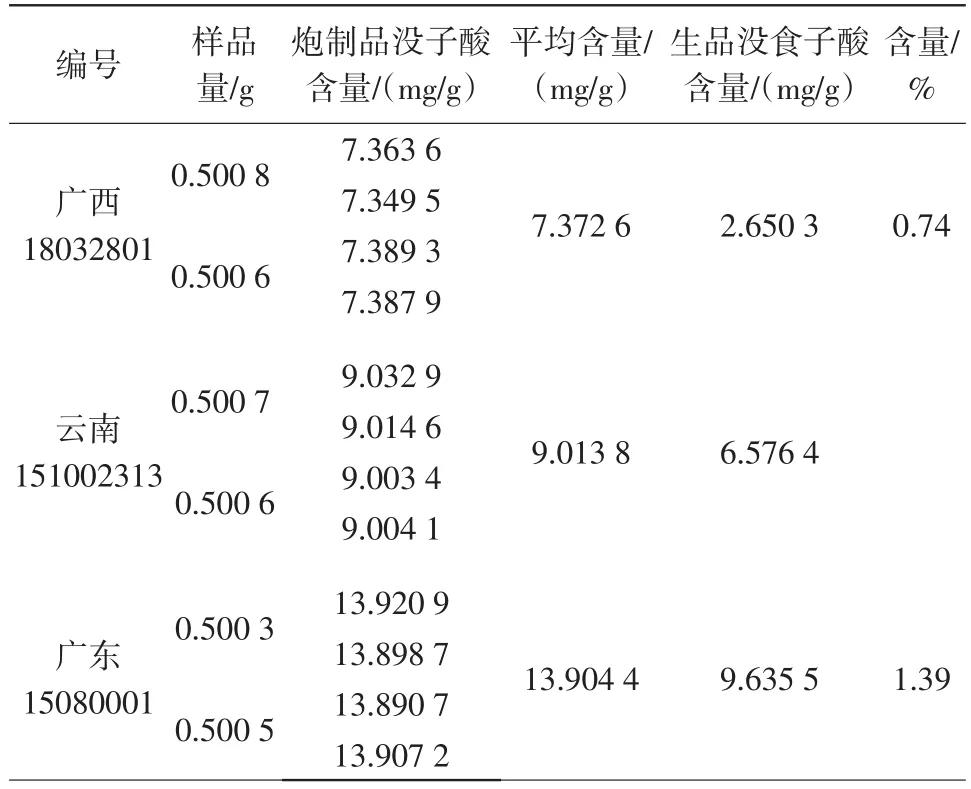
表9 不同批次煨诃子中没食子酸含量测定结果
结果显示,3批次供试品中没食子酸平均含量为0.74%~1.39%,均比诃子原药材的没食子酸含量有所提高,考虑到药材的采收时间、地点不同等因素,拟定为本品按干燥品计算,含没食子酸(C7H6O5)不得少于0.74%。
3.2 含量测定本研究对煨诃子的提取方法(超声提取、加热回流提取)、提取时间(20 min、30 min、40min)及提取溶剂(甲醇、70%甲醇、50%甲醇)进行了选择。结果显示,以甲醇为提取溶剂,超声提取30 min时效果最佳。
本研究对煨诃子的水分、总灰分、浸出物、显微鉴别、薄层鉴别、及含量测定等项目进行较为系统和全面地研究。其测定方法简便准确,方法学考察表明精密度、重复性、稳定性、加样回收率的范围均符合要求,故为蒙药饮片煨诃子的质量控制提供了有力的实验依据。
猜你喜欢
河北农业(2022年8期)2022-09-28 06:59:32
山东陶瓷(2021年5期)2022-01-17 02:35:46
陶瓷学报(2021年1期)2021-04-13 01:33:08
中国林副特产(2019年6期)2019-01-11 02:33:06
中成药(2018年1期)2018-02-02 07:20:14
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:41
西藏科技(2016年9期)2016-09-26 12:21:39
海峡科技与产业(2016年3期)2016-05-17 04:32:14
云南中医学院学报(2015年3期)2015-07-31 18:09:28
山东医药(2015年14期)2015-04-04 14:00:07
