
PhIP化学模型体系中前处理及UPLC-MS检测方法的研究
2019-08-28 12:27:56陈妍方赵月亮张娜娜范大明李利洁赵建新
食品工业科技 2019年15期
陈妍方,赵月亮,张娜娜,范大明,,*,李利洁,赵建新,5,张 灏,5
(1.江南大学,食品科学与技术国家重点实验室,江苏无锡 214122;2.江南大学,国家功能食品工程技术研究中心,江苏无锡 214122;3.江南大学食品学院,江苏无锡 214122;4.香港大学生命科学学院,香港薄扶林 SAR;5.上海海洋大学食品学院,上海 201306)
在高温烹调过程中,肉制品中的氨基酸、肌酐和葡萄糖三种成分会产生杂环胺,是一类具有致癌、致突变性的多环芳香族化合物[1]。其中,2-氨基-1-甲基-6-苯基-咪唑[4,5-b]吡啶(PhIP)是高温处理肉类产品时生成的含量最丰富的杂环胺。1999年,国际癌症研究机构将PhIP划为2B类潜在致癌物[2],Sinha等人的研究表明,摄入PhIP增加了消费者的患癌风险,其摄入量与患病率存在正相关关系[3-4]。随后,美国健康和人道服务部将PhIP列为合理预期的人类致癌物质,并将其列入国家毒理学计划[5]。因此,需要对烹调肉制品中产生的PhIP进行定量分析,为相应抑制手段的探究提供依据。
由于肉类制品中含有大量的油脂、蛋白质等物质,体系本身十分复杂,且其中的PhIP含量属于ng/g水平,故需要对其进行复杂前的处理,才能进行后续的检测和分析[6]。食品体系的复杂性给PhIP的相关的反应机理探究增加了困难,为了减少干扰,Shioya等人[7-8]首次简化了实验对象,采用氨基酸、肌酸和葡萄糖三种PhIP的前体物质组成了化学模型,研究其形成机制。化学模型体系为反应机理层面的研究排除了很多干扰,被广泛采用[9-10]。
目前,化学模型体系的前处理多采用液液萃取结合SPE固相萃取的方法,萃取后收集反应液,过固相萃取小柱后氮吹。但此方法的操作精度要求较高,也会有洗脱不净的情况出现,均会直接导致测得的PhIP含量偏低。复杂的处理步骤会增大体系中PhIP损失的概率,也会降低实验的可重复性和回收率[11-12]。本文在传统PhIP检测方法的基础上,建立了一种不需要固相萃取小柱的样品处理方法。此方法将会简化操作过程,提高可重复性,可作为一种特定的检测化学体系中PhIP的新型方法。
1 材料与方法
1.1 材料与仪器
苯丙氨酸、葡萄糖、肌酸酐、乙酸乙酯、NaOH 国药集团化学试剂有限公司;甲醇、乙酸铵 江苏博美达生命科学有限公司;2-氨基-1-甲基-6苯基咪唑并[4,5-b]吡啶(PhIP) Toronto Research Chemicals(Toronto,加拿大)。
Thermo HAAKE AC200-S13油浴锅 美国Thermo公司;79480-3冷冻干燥系统 CONCO公司;Waters超高液相色谱-三重四级杆串联质谱仪(UPLC-TQD) 美国Waters公司;ACQUITY BEH C18(1.70 μm,50 mm×2.10 mm)、ACQUITY BEH T3(1.70 μm,50 mm×2.10 mm)、ACQUITY BEH phenyl(1.70 μm,50 mm×2.10 mm)色谱柱 美国Waters公司;QSC-12T氮吹仪 泉岛公司。
1.2 实验方法
1.2.1 标准溶液的配制 参考蒋智等[6]的方法,精确称取10.0 mg的PhIP标准品,用甲醇进行溶解,配成浓度为100 μg/mL溶液。将标准液用甲醇进行梯度稀释,配制成2.5、5、10、20、50、75 mg/mL的标准溶液。
1.2.2 PhIP化学模拟体系的建立 本研究中的化学模型以李利洁等[13]的方法为参考并进行调整。将20 mmol/L肌酸酐、10 mmol/L葡萄糖、20 mmol/L苯丙氨酸溶于蒸馏水中,配成100 mL的反应体系。在有配垫片的螺口玻璃瓶中加入10 mL反应液,拧紧瓶盖,设定油浴锅加热条件为128 ℃、油浴2 h,冷却至室温。
1.2.3 样品前处理 取1.2.2中油浴加热后的反应溶液于冷冻干燥机中冻干,冻干程序为:0~4 h,-40 ℃;4~19 h,-30 ℃;19~34 h,-10 ℃;34~38 h,10 ℃;38~42 h,20 ℃。以冻干前反应液的体积为依据,每10 mL反应液得到的冻干样品中加入2 mL的2 mol/L NaOH和2~8 mL乙酸乙酯,形成碱性萃取环境。将萃取体系震荡3 min,超声处理4~12 min,6000 r/min离心5 min,取上清液氮吹,去除溶剂,加入0.20 mL甲醇震荡溶解,用0.22 μm有机滤膜进行过滤,用超高效液相色谱串联四级杆质谱法(UPLC-MS)进行PhIP含量测定。
1.2.4 化学模型体系中PhIP的UPLC-MS检测条件
1.2.4.1 液相色谱条件 参照李利洁等[13]的方法对液相条件进行调整,色谱柱为Waters BEH phenyl柱(50 mm×2.10 mm,1.70 μm),流速为0.30 mL/min,设定柱温为35 ℃,进样量为10 μL。流动相为:乙腈(A),0.1%甲酸水溶液(B),梯度洗脱程序为:0~8 min,10% A;8~9 min,70% A;9~9.5 min,10% A。在下次进样前,用10% A液保持5 min,用于平衡柱子。
1.2.4.2 质谱条件 参照蒋智等[6]的方法对质谱检测条件进行优化。离子源为ESI+,采用多反应监测模式进行检测;设定离子源温度为120 ℃;喷雾电压2.8 V;毛细管电压为2.50 kV;脱溶剂温度400 ℃;脱溶剂气体流速700 L/h;锥孔气体流速50 L/h;碰撞气体流速0.30 mL/min。
1.3 数据处理
所有实验均重复三次,结果数据用平均值±标准偏差的形式表示,数据处理采用Microsoft Excel 2010;图表绘制采用软件GraphPad Prism;显著性分析采用软件SPSS Statistics;LC-MS图谱分析采用软件Masslynx V4.1。
2 结果与讨论
2.1 前处理条件的选择
已有较多研究对不同样品体系中的PhIP前处理方法进行了优化。Calbiani等[16-18]不断地改进固相萃取方法的萃取剂种类、流动相以及柱条件等,提高了分析的可重复性和稳定性。但Khan等[19]认为固相萃取法溶剂量很大且净化步骤复杂,故采用加压流体进行提取,使杂环胺的回收率高达62%。本研究提出了一种无需使用固相萃取柱的前处理方法,由于PhIP可溶于蒸馏水和有机溶剂,故先采用冻干法对将大体积的反应体系浓缩,又可同时去除体系中的水分,使得PhIP更容易被萃取进入乙酸乙酯层。随后,直接收集乙酸乙酯层溶液,进一步氮吹浓缩,使用甲醇复溶,过膜后待测。
2.1.1 提取溶剂及用量的选择 提取杂环胺常用的溶剂主要有:乙酸乙酯[20-21]、二氯甲烷[22]、二氯甲烷(含5%甲苯)[23]等。本方法中浓缩萃取后的反应液需要取上层液体氮吹,但二氯甲烷密度大,涡旋混合离心后萃取效率差,而采用乙腈回收率较低,且不易用氮气吹干,故选择密度小且易于氮吹的乙酸乙酯作为萃取溶剂。由于PhIP的疏水性和亲水性取决于所处介质的pH,随着pH升高,疏水性增强,PhIP更易溶解于有机提取溶剂中[24]。为了提高萃取率,本方法向乙酸乙酯中加入2 mol/L NaOH构成碱性环境,以促进萃取。在10 mL的化学模型反应体系中,固定NaOH用量,考察乙酸乙酯用量(2、3、4、5、6、7、8 mL)对PhIP提取效果的影响。图1为乙酸乙酯用量对PhIP提取量的影响。如图1可知,提取到的PhIP浓度随乙酸乙酯使用量的加大,呈现上升的趋势,但当提取剂用量大于5 mL时,PhIP浓度随萃取剂用量缓慢上升 。此结论与肖正华等[25]结果一致,结合后续浓缩步骤与成本综合考虑,选取每10 mL的反应溶液进行前处理,以加入5 mL乙酸乙酯为宜。

图1 乙酸乙酯用量对PhIP浓度的影响
2.1.2 萃取时间的选择 取10 mL反应液,加入5 mL乙酸乙酯和2 mL NaOH,振荡萃取4、6、8、10、12 min后,测定PhIP的萃取效果,结果见图2。发现随萃取时间的延长,PhIP浓度先增加后减少,在10 min时提取量达到峰值。此结果与邓燕莉等[26]的结论一致,体系中非PhIP的杂质被萃取的量随着萃取时间的延长而增加了,导致了PhIP的纯度下降。所以萃取时间选择为10 min。通常,采用固相萃取法进行样品前处理时,萃取时间长达2~3 h[6]。与固相萃取法相比,本法将萃取时间缩短为10 min,对溶液进行简单快捷又有效地萃取,简化了实验。

图2 萃取时间与PhIP提取量的关系
2.2 质谱条件的确定
由于化学模型体系中的PhIP属于痕量物质,对PhIP的检测采用串联三重四级杆质谱仪进行。通过针泵注射1 μg/mL标准液进样,由于被测物质PhIP结构中含有氨基和亚氨基的电离特性,检测采用ESI+模式。蒋智等[6]的实验表明,离子强度随毛细管电压的升高而逐渐增强,在电压达到2.5 kV时,离子强度趋于稳定;在较低的碰撞能量下,能量不足,导致得不到PhIP的碎片离子,当锥孔电压调节到35 V时,可以获得稳定的分子离子m/z 225。对PhIP的母离子进行全扫描可得到子离子,进入二级质谱后受到碰撞能量的作用,失去-CH3会形成质荷比为210.1的离子,失去-HCN基团形成质荷比为183.1的离子,这两个离子是PhIP的特征离子,选择信号最强的225.1>210.1作为定量离子。在多反应监测模式下,能够通过筛选目标物的特征离子来定性定量,可以提高检测方法的灵敏度和选择性。

图3 PhIP的碎片离子质谱图
2.3 PhIP检测色谱条件的确定
2.3.1 不同色谱柱对分离效果的影响 实验中采用BEH C18、BEH T3和BEH phenyl三种分离原理不同的色谱柱对样品体系中的PhIP进行分离,PhIP的信号强度来对色谱柱进行筛选,结果如图4所示。由图4可知,BEH C18、BEH T3、BEH phenyl三种色谱柱得到的PhIP保留时间分别为3.44、3.49、4.49 min。但是使用BEH C18柱进行分离时,拖尾现象非常明显。BEH T3柱的拖尾不明显,色谱峰却有轻微分叉。C18柱和T3柱分离效果不佳,是由于这两种色谱柱只能在反相UPLC条件下分离和保留极性有机化合物,对PhIP这样的极性杂环胺分离效果较差。而采用BEH phenyl柱得到的峰型对称性好,没有分叉现象。这与满正印等[27]的结论相似,可能由于苯基柱能形成p-p共轭相互作用,对极性芳香族化合物的保留能力比C18柱和T3柱强。此外,苯基柱对含有苯环的目标物分离更有选择性,更利于PhIP的分离[27]。因此,最终选择BEH phenyl柱进行PhIP的色谱分离。
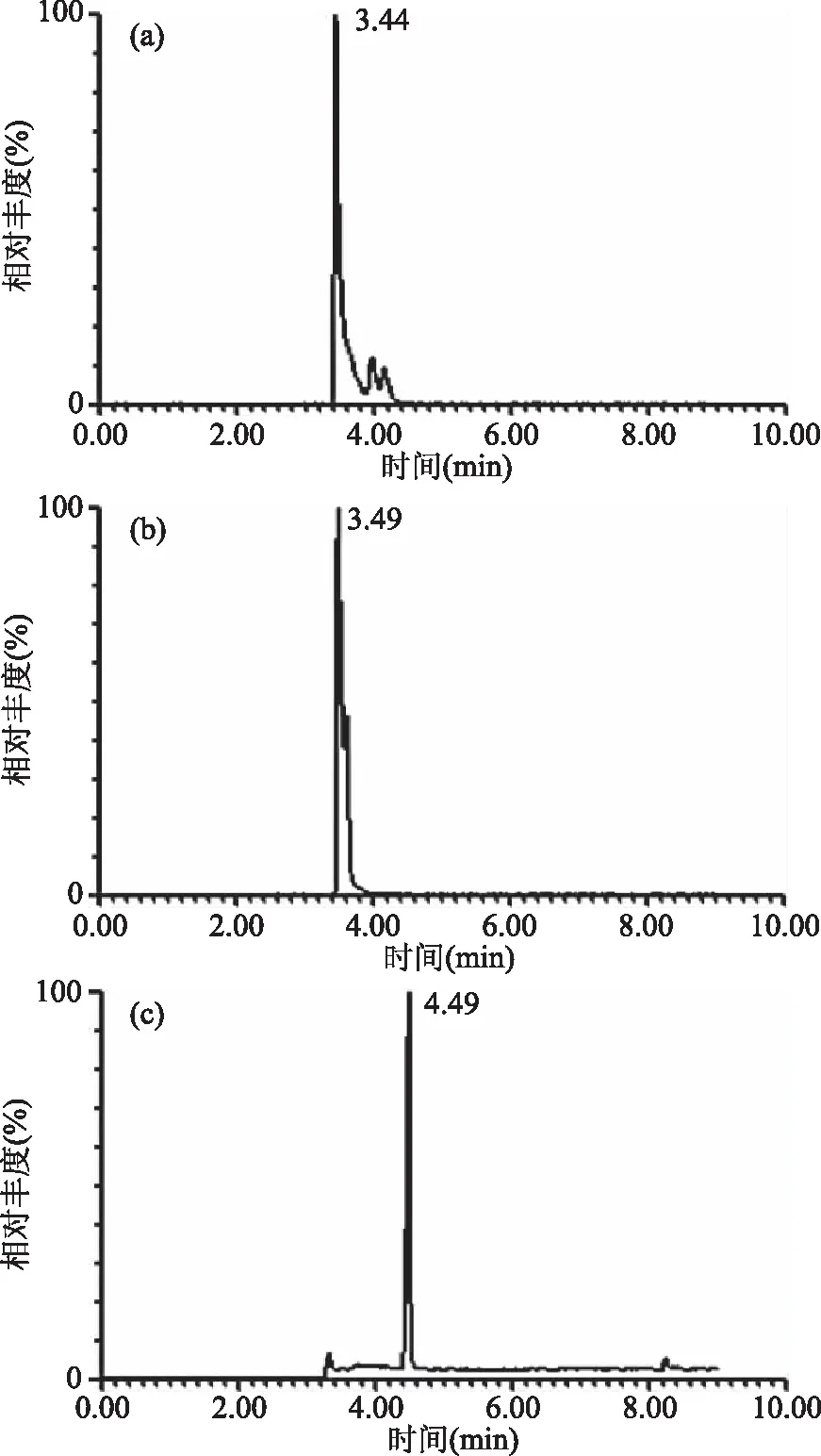
图4 采用不同色谱柱——BEH C18(a)、BEH T3(b)、BEH phenyl(c)对PhIP的分离效果影响
2.3.2 流动相对分离效果的影响 本研究固定乙腈为流动相A,分别考察了0.1%甲酸水和3 mmol/L乙酸铵为流动相B对PhIP的分离效果。Shah等[24]采用乙酸铵作为流动相,因其能使峰型得到改善,溶剂峰小,降低干扰性。乙酸铵容易残留在柱子中,对色谱柱损伤较大,且成本高,价钱昂贵。仲伶俐等[28]采用选择phenyl色谱柱测定水果中萘衍生物,发现用乙腈-0.1%甲酸作为流动相能够改善色谱峰形,且对保留值并无影响,故本实验对比了0.1%甲酸水和乙酸铵分离PhIP的效果,结果如图5所示。由图5可知,采用0.1%甲酸水作为流动相B时,缩短了保留时间,且基线比采用乙酸铵时稳定,峰型较好。0.1%甲酸能提供质谱离子化时所需的H+,促进分子的电离,保证化合物在质谱下的离子化,大大增强检测信号的强度[29-30]。故本研究采用乙腈-0.1%甲酸水作为流动相。
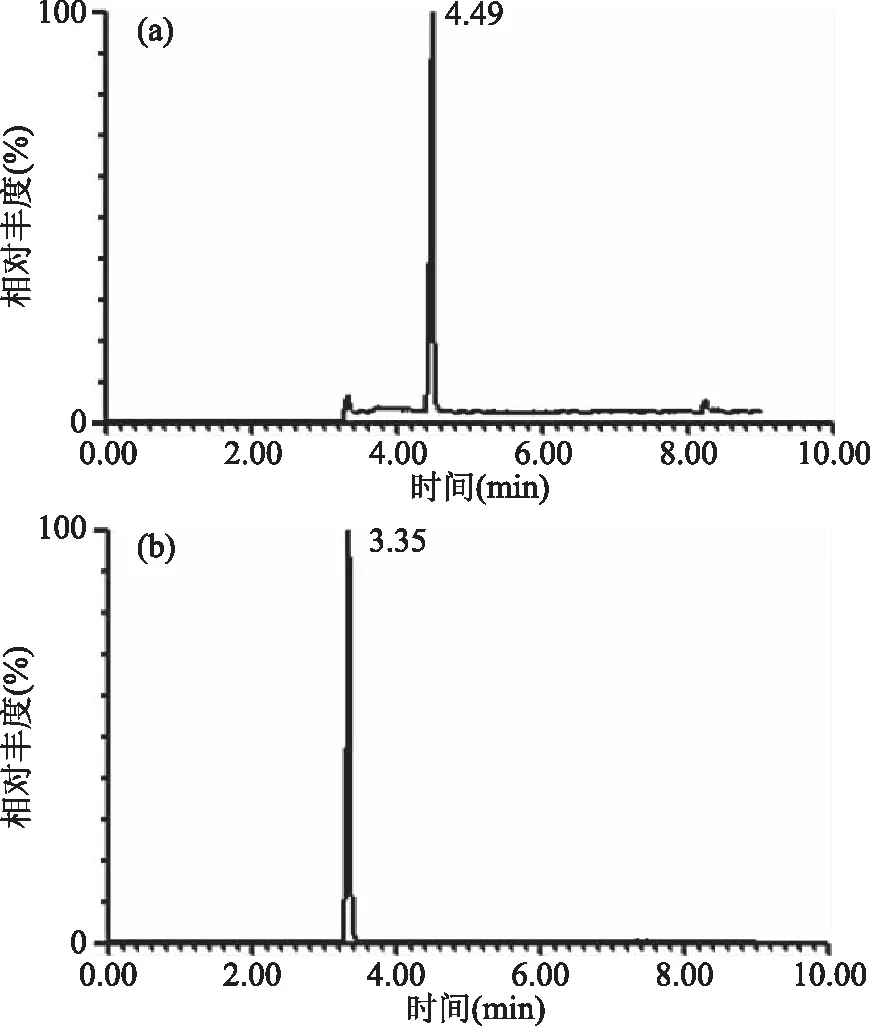
图5 采用流动相3 mmol/L乙酸铵(a)和0.1%甲酸水(b)对PhIP的UPLC-MS/MS分离效果的影响
2.4 样品前处理与检测方法的评价
2.4.1 线性范围、检出限和定量限 以对应的标准液浓度为X轴,PhIP峰面积为Y轴绘制标准曲线,其线性关系和相关系数见表3,结果显示,本方法的线性范围为0.5~15 ng/mL,决定系数为0.999。添加标准溶液后,测得的PhIP定量限为0.332 ng/mL,检出限为0.110 ng/mL。

表3 冻干法应用于PhIP分析的方法学数据Table 3 Methodology data for the freeze-drying method applied to the analysis of PhIP
2.4.2 回收率和精密度 向空白样品中添加10、15、30 ng/mL三个浓度的PhIP标准品,测定的平均回收率见表4。加标水平为10、15、30 ng/mL时,新方法的平均收率分别为78.16%、81.22%、82.03%,试验中检测PhIP的相对标准偏差为6.32%、5.40%、4.28%,实验结果表明,冻干萃取法的稳定性和可重复性均较好。

表4 冻干法应用于PhIP分析的平均回收率实验结果Table 4 Mean recoveries of freeze-drying method applied to the analysis of PhIP
3 结论
本研究对化学体系中PhIP的前处理和UPLC-MS检测方法进行了优化,重点研究了浓缩方式、萃取剂用量、萃取时间三个因素对PhIP提取效果的影响。将样品冻干处理后,采用NaOH-乙酸乙酯2∶5的比例进行萃取,可使PhIP得到比较完全的萃取。采用Waters BEH phenyl色谱柱,以乙腈-0.1%甲酸为流动相进行梯度洗脱,可以得到对称性好的峰型。本方法检测PhIP的线性范围为0.5~15 ng/mL,决定系数为0.999,检出限为0.110 ng/mL,定量限为0.332 ng/mL,在加标水平为10、15、30 ng/mL时的平均收率分别为78.16%、81.22%、82.03%,相对标准偏差(RSD)分别为6.32%、5.40%、4.28%。本方法对样品前处理过程和检测条件进行了优化,可用于分析化学体系中的PhIP含量。目前国内对于化学体系中PhIP检测方法的报道还很少,本法的建立为探究化学体系中PhIP的有效抑制方法提供了检测依据。本方法操作简便、可重复性高,可作为测定化学模型中PhIP的有效方法。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
中成药(2018年6期)2018-07-11 03:01:04
中国蜂业(2018年4期)2018-05-09 06:25:08
中成药(2018年4期)2018-04-26 07:12:47
中成药(2017年8期)2017-11-22 03:19:25
中国兽医杂志(2016年7期)2016-08-30 01:08:28
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
当代化工研究(2016年6期)2016-03-20 16:21:46
食品工业科技(2014年23期)2014-03-11 18:19:04
无机化学学报(2014年3期)2014-02-28 17:30:58
