
六倍体小麦基因组注释流程构建与优化①
2019-08-22 02:31祝海栋李瑞琳何小雨韩鑫胤牛北方
计算机系统应用 2019年8期
祝海栋, 李瑞琳, 何小雨, 赵 丹, 韩鑫胤, 牛北方
(中国科学院 计算机网络信息中心,北京 100190)
(中国科学院大学 计算机与控制学院,北京 100190)
小麦生产对保证粮食安全和农业可持续发展具有重要的现实意义,促进小麦的增产和品质改良成为当前小麦育种研究的前沿热点. 为了培育具有优良性状的新品种,首先要定位控制目标性状的基因,因此建立一套完整准确的大尺度基因组注释流程成为培育新品种过程中的难点之一. 基因组注释主要包括基因识别和基因功能标注两个方面[1],本文的主要研究方向是基因识别,主要目标是准确定位基因位置及发现物种特异性基因.
近些年,基因组测序技术突飞猛进,其发展过程包含三个阶段:1975年由桑格和考尔森开创的链终止法标志着第一代DNA测序技术的诞生,但测序成本高、通量低等缺点严重影响了其大规模的应用; 第二代测序建立在聚合酶链式反应扩增的基础上,主要特点是为边合成边测序,测序结果读长短、测序速度快、吞吐量大[2]; 第三代测序技术的核心是以单分子为目标,旨在解决第二代测序在准确性和组装困难方面的问题.测序技术的高速发展,大大满足了测序深度、重测序等大规模基因组的研究需求,改变了生命科学诸多领域的研究面貌,也给小麦等大尺度基因组的注释研究奠定了重要基础.
1 小麦基因组注释流程研究现状
传统的基因注释方法主要为数据库比对,通过把基因组片段与已有的亲缘物种基因数据库比对,得到目标基因. 这种方法较为简便,但具有三个明显的缺点:一是对比速度慢,原因是该方法中需要与较多的数据库进行比对分析,因此耗时长,尤其是用于小麦等较大基因组时该缺点更为明显; 二是难以发现新的基因,由于依赖数据库比对得到的基因都是目前相近物种中广泛存在的基因,物种特有的基因不会被识别,造成注释的不完整. 转录组测序可以全面快速的获取物种在某一时期和特定组织中所有表达的基因序列,常被用于研究物种基因结构和基因功能[3]. 但是转录组分析软件繁多,缺乏统一的选择标准,且分析过程中涉及多个软件配合完成,分析流程中不可避免地会存在软件间衔接困难、格式转换和大量数据重复读写等问题. 另外,由于各种软件在内存、CPU等资源利用方面存在较大差异,且多数情况下生物信息学中的分析过程依赖于脚本生成的流程,没有并行优化,因此资源利用率和分析效率较低. 针对上述问题,本文提出了整合基因组和转录组数据进行基因注释的分析流程,以提高注释的完整性和准确性.
2 实验数据与测试环境
本次研究中使用的测试数据包括:科农9204小麦基因组组装数据,数据大小14.24 GB; 二代转录组测序样本77个,单样本大小约为17 GB; 三代全长转录组测序样本2个,单样本大小约为40 GB. 测试环境为超级计算系统“元”. 其包含270台计算节点,每节点采用2个Intel Xeon E5-2680V3处理器(2.5 GHz、12核),单节点CPU计算能力 0.96Tflops,配备256 GB内存.操作系统为Linux version 2.6.32-358.el6.x86_64,CentOS release 6.4 (Final). 系统中配置Python、Perl、C++等基本编译和运行环境.
3 基因组注释分析软件流程
本节分为3个部分,建立小麦基因组注释分析流程,并对部分环节实现优化.
3.1 数据库比对注释
数据库比对注释是最传统和最常用的注释方式.其主要方法是把待注释的基因组逐一与各个近亲物种已有基因比对,获取注释结果. TriAnnot[4]是为解读小麦基因组而开发的一个流程,集合了Blast、Repeat Masker等开源软件,比对了NCBI[5]、TAIR10[6]等开放数据库,对转座子、编码基因、非编码序列、分子标记进行了多步的处理分析,可以得到比较完整的注释结果. 因此,本文对TriAnnot注释软件进行优化并对科农9204小麦基因组进行初步注释.
3.1.1 优化方法
为了提高注释效率,本研究的主要贡献是实现TriAnnot注释软件的优化,重点分为3个方面:单任务多实例并行优化、多核计算并行优化,多数据库查找并行优化,下面给出具体的方法与实现.
首先,对TriAnnot注释软件的单任务多实例并行优化. 六倍体小麦基因组较大,每条染色体的平均长度接近700 MB,给序列比对带来许多困难. 为了便于比对分析,在注释过程中,必须把染色体切分成小的片段,本研究中选择的切分大小为1 MB,切分时的保留的重复长度为50 KB. 切分后的每个片段即为每个实例,实例之间相对独立. 为了提高注释速度,本研究采用了多实例并行,即在每个时刻都有多个实例同时执行. 因为每个步骤的CPU和内存使用率各不相同,该优化策略可以实现资源的充分利用.
其次,实现TriAnnot注释软件的单任务多实例并行优化. 在实验中,针对每个软件的特点,本研究采用相应的调度方式优化. RepeatMasker通过相似性比对来识别重复序列,可以屏蔽序列中转座子重复序列和低复杂度序列[7]. 本研究在流程中加入了RepeatMasker的多核心并行,可以根据机器硬件情况指定4至24核心实现并行运行,并且可以通过使用-qq指令加快比对效率.
最后,实现对TriAnnot注释软件的多数据库查找并行优化. SIMSearch软件通过使用多个同源数据库进行序列比对找到亲缘关系较近的基因序列. 为了加快同源基因的查找速度,研究采用了多数据库并行的方案,把多个同源数据库同时读取到内存中,将每个基因片段在多个核心上与不同的数据库进行对比.
3.1.2 软件与数据库
TriAnnot依赖的软件及其下载地址如表1所示,数据库及其下载地址如表2所示.

表1 TriAnnot主要依赖软件

表2 TriAnnot主要数据库
3.1.3 分析流程
该步骤的输入为基因组装得到的KN9204小麦基因序列文件. 六倍体小麦有21条染色体,此外还有少量未有效定位到染色体上的基因片段,在其中加入100个未知碱基标识“N”,构成未分组染色体,共22条fasta序列,每条序列单独输入.
TriAnnot软件运行前需要下载完整的基因数据库.主要参数包括:-W指定工作目录,-s指定输入的fasta文件,-t指定注释流程xml文件,--type设置输入为核酸,--maxlength设置最大序列长度,--splitseq设置超过最大长度的序列自动切分,--overlap设置切分时冗余长度.
软件的输出为gff文件,包含了详细的内含子、外显子、编码区、转座子等注释.
3.2 转录组高通量测序
为了准确注释物种特异性基因,本研究结合了转录组高通量测序数据,选取了苗期、孕穗期、7天、14天等不同时期的根、叶、穗等不同组织的样本,测序深度约为30 X. 常用的转录组分析工具有HISAT、SATR、StringTie、Cufflinks等. 使用不同的分析工具和方法对分析结果的准确度和耗时影响较大,需要根据特定的数据集及特定的研究目标选择合适的分析工具和方法. HISAT解决了转录组中仅有不连续的外显子难以比对的问题,对比上代主流转录组比对工具Tophat效率高50倍,且内存需求更少[8]. StringTie继承于Cufflinks,在准确性方面有了较大提升,且可以通过输入数据库比对注释结果提高在已知基因区域的准确性,在组装的过程中会计算每个基因及可变剪切的表达水平. 综合以上优点,对于复杂的小麦基因组,本文使用HISAT[9]和StringTie[10]工具进行转录组组装.主要分为以下四个步骤
(1) 建立HISAT2基因组索引. 转录组数据分析过程遇到的第一个问题就是,小麦上亿条reads如何在保证错误率在可接受的范围内,高效率地比对到基因组上. 针对上述问题,需要根据基因组序列使用hisat2-build命令建立索引.
(2) 将所有二代测序reads比对到基因组. 使用HISAT2利用基因组索引将高通量测序reads比对到基因组上. 参数-p指定并行核心数,-x指定索引位置,--dta为组装提供锚点. 使用samtools将比对结果按染色体和起始位点排序.
(3) 使用StringTie对排序完成的reads进行组装.不同组织中表达数据差异相对较大,比对到基因组的reads也各有不同,这些因素都会影响组装的效率.
(4) 将所有转录本的组装结果使用StringTie的merge模块合并. 由于不同组织和不同时期表达的基因各不相同,为了获取更加完整的注释,需要对多个测序样本合并. merge步骤可以跨多个测序样本生成统一的转录本. 首先要创建一个文本文件,该文件包含所有转录本组装结果路径,文本的每行是单个样本组装结果文件路径. 参数设置为:--merge指定使用合并模块,-p指定并行核心数,输入上述文本文件,即可得到最终的二代转录组组装结果.
3.3 全长转录组单分子测序数据处理
二代测序可以准确地进行基因定量分析研究,但是受读长限制,不能得到全转录本的信息. 全长转录组采用单分子实时测序技术,通过构建哑铃型文库,以环形方式循环测序[11]. 因此,通过全长转录组单分子测序可以不经过组装,准确、直接地获取整个转录本. 三代测序存在单碱基错误率较高的问题[12],本研究使用PacBio公司发布的SMRTLINK Pipeline[13],对三代测序得到的数据进行过滤与质量控制. 由于全长转录组测序成本相对较高,本次研究采取了常用的组织混合测序方式. 选取了叶、穗、幼叶、幼根四种组织混合,设置两个生物学重复,共得到两组测序数据. 数据处理过程主要分为以下3个步骤:
(1) 使用SMRTLINK进行三代测序数据的清洗.主要分为三个步骤,首先召回环形一致性序列,包括单碱基纠错和序列过滤; 然后对序列分类,包括去除接头、polyA尾部和串联子; 最后进行迭代的聚类纠错,主要是合并相似的序列,形成全长转录本. 该软件提供了用户可视化接口,安装后使用浏览器访问服务器地址的对应端口即可进入管理界面. 在管理界面中,使用“数据管理”选项导入原始测序结果文件,然后使用“SMRT分析”选项,选择分析流程为“Iso-Seq”,设置相关参数,选取对应的样本即可开始全长转录组的纠错.在本次研究中,我们设置的参数主要有以下几个:By Strand CCS:OFF; Maximum Dropped Fraction:0.8;Maximum Subread Length:15000; Minimum Predicted Accuracy:0.75; Minimum SNR:3.75; Polish CCS:ON;
其余参数均为默认值.
(2) 使用GMAP[14]比对全长转录本到基因组.GMAP具有一次对多条reads同时进行比对的优点,比对结果较为可靠,因此,本文采用GMAP将全长转录本比对到基因组上. 为了提高运算速度,GMAP比对阶段对全长转录本序列进行数据分割,将分割后的多个数据进行并行处理. 首先使用gmap-build建立索引,由于小麦基因组较大,会自动使用长索引. 使用-D参数指定索引存储位置,-d参数指定索引前缀,输入基因组fasta文件即可开始建立索引,然后使用gmapl命令开始比对. 指定的索引存储位置和前缀需与上述过程中对应参数相同,-B指定批处理个数,-t指定并行核心数,-f指定输出格式,-O指定顺序输出. 使用samtools将bam文件按染色体和起始位点排序.
(3) 合并多个样本的全长转录组结果. 合并时使用TAMA软件,共分为两个步骤. 首先根据比对到基因组上的位置情况合并可变剪切,然后合并多个测序样本的转录本.
3.4 合并注释结果
为了得到高质量的注释结果,需对上述结果进行合并和过滤,在本次研究中我们开发了一个自动化合并注释的软件Annotator,该软件包含的功能模块有格式转换,结果合并,去除重复序列,过滤可变剪切,根据证据支持评价可信度,编码区预测,蛋白翻译等多个步骤,最终生成gff注释文件和转录本序列、编码区序列、编码蛋白序列. Annotator详细流程如图1所示. 合并过程分为以下5个步骤:
(1) 转换结果文件为bed12格式. 本步骤调用了cufflinks[15]软件的gffread模块和bedops[16]软件,将数据库比对注释得到gff文件和二代转录组组装得到的gtf文件转换为bed文件. bed格式使用单行定义单个基因,具有简单易读的特点.
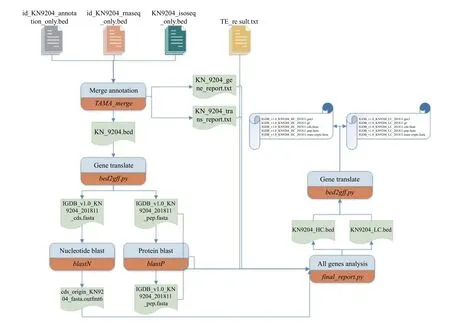
图1 注释合并流程
(2) 合并数据库比对注释、二代转录组组装、三代全长转录组结果. 本步骤使用了TAMA的merge模块,生成含有全部基因的bed文件. 根据每个基因的支持证据的不同,分为高可信度基因和低可信度基因.
(3) 过滤重复的可变剪切. 由于测序误差或reads组装错误的不可避免,测序结果中可变剪切会出现许多冗余,因此需要对重复的可变剪切进行过滤. 过滤过程中,保留的优先级依次为全长转录组得到的可变剪切结果、数据库比对注释结果中的可变剪切,由于二代转录组组装有更多的错误可能,其优先级最低.
(4) 预测所有基因的编码区. 该步骤使用三种可能的翻译方式分别将基因翻译为氨基酸序列,取最长的序列,得到基因的编码区.
(5) 翻译编码区序列. 根据注释结果中的编码区位置,将核酸序列翻译为氨基酸序列,生成序列文件.
4 实验结果与分析
经过优化,使用TriAnnot注释科农9204基因组的重复序列时,速度提升达到60%,在1号染色体上的测试结果如图2所示.
在转录组高通量测序过程中,建立索引过程输入全基因组大小约为14 GB,耗时为8122秒,最大内存使用约为144 GB. 序列比对时,输入共77个样本,双端测序的单个fastaq文件大小约17 GB,比对耗时约640秒. StringTie组装输入bam文件大小约为7 GB,耗时在10小时至24小时不等.三代全长转录组测序中单样本bam文件为38 GB,运行时间约为149小时. 最终分别输出高质量和低质量的全长转录组fasta序列. 最终得到的环形一致性序列质量分布如图3所示,超过50%的序列质量均在0.99以上,可信度较高.

图2 注释耗时变化
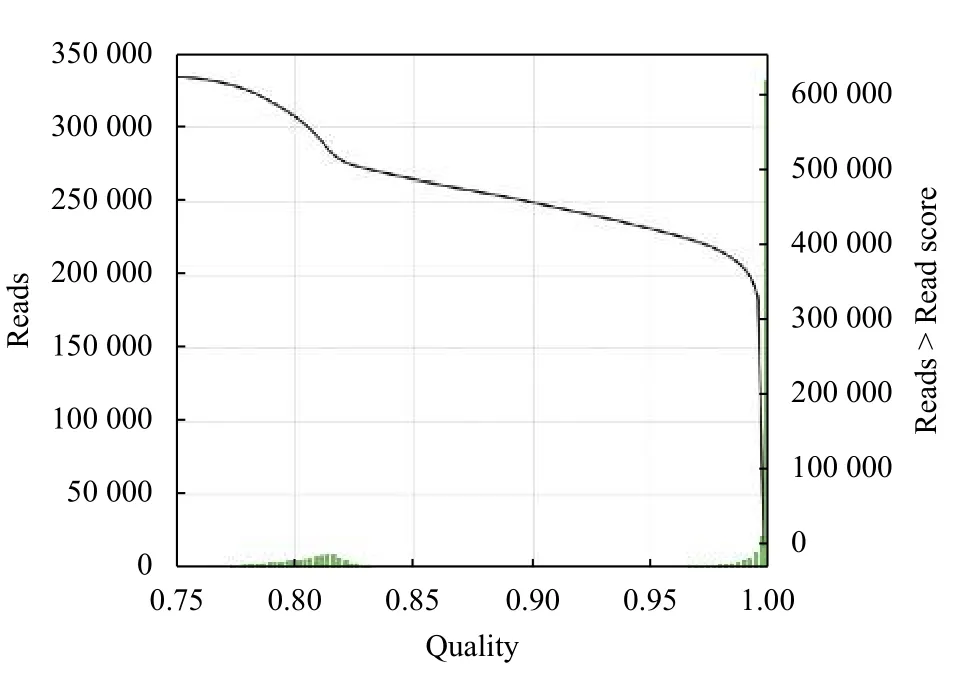
图3 全长转录组质量分布
本流程在六倍体小麦科农9204基因组上完成测试,共注释出110 326个高可信度基因. 对比同源的中国春小麦基因组,其注释包含107 891个高可信度基因[17],其中有102 413个基因匹配,占中国春基因总数的94.9%,占科农9204基因总数的92.8%,具有高度一致性,这说明了本流程注释结果具有较高的准确性.
5 结论与展望
本文提出了一种综合运用数据库比对、二代转录组高通量测序、三代全长转录组测序技术获得准确注释的分析流程,并独立研发了注释软件Annotator. 随后对流程中用到的部分软件进行了优化,大大提高了注释效率,为大尺度多倍体基因组提供了一个较为成熟的注释软件流程.
当前流程也存在一些问题:(1) 注释速度仍然较慢.数据库比对注释过程是性能提升的主要瓶颈,仍需优化. (2) 成本较高. 注释的准确性依赖于较高的测序深度,这会带来成本的大幅提高,尤其是三代测序更为如此,这大大限制了该流程的广泛应用.
因此,在未来的工作中将尝试解决上述问题,以进一步优化整个流程. 针对注释速度问题,可以对整个基因组进行更细粒度的并行处理,提升比对过程中的并行效率; 此外可以使整个比对过程均在内存中进行,避免中间结果写入硬盘,减少不必要的时间开销. 针对注释中测序成本问题,可以在成本较高的三代全长转录组测序中采取多组织混样测序的方案,选取最关注的组织和时期的样本混合,通过单次测序降低成本.
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
今日农业(2021年14期)2021-10-14
今日农业(2021年11期)2021-08-13
今日农业(2021年10期)2021-07-28
科学导报(2021年29期)2021-06-03
ViVi美眉(2019年8期)2019-09-10
科海故事博览·下旬刊(2019年6期)2019-04-16
劳动保护(2018年5期)2018-06-05
