
非晶态Fe0.51P0.49合金薄膜的电化学制备及其性能研究
2019-08-13 07:11荣文倩郑小美张朋越葛洪良
中国计量大学学报 2019年2期
荣文倩,郑小美,张朋越,葛洪良
(1.中国计量大学 材料科学与工程学院,浙江 杭州 310018;2.中国计量大学 标准化学院,浙江 杭州 310018)
全球变暖以及日益严峻的环境污染,促使我们必需使用和开发绿色能源和可再生能源,如潮汐能,风能和太阳能[1-2]。由于这些自然资源的不可连续使用性,开发应用于智能电网领域的高比容量能量存储系统是一项当前紧迫的任务。目前,可充电锂离子电池(LIBs)作为主要的储能设备,广泛应用在便携式电子设备、电动汽车、人工心脏起搏器和植入式电子医疗设备[3-4]。为了满足智能电网和电动汽车所需的高能量密度和安全性能的要求,很多科学家集中研究开发新的电极材料。在负极方面,目前商业化的石墨负极,由于其低的理论比容量为372 mAh·g-1,而限制了它在智能电网和电动汽车等方面的应用。因此,开发具有高可逆容量和长循环寿命的的锂离子电池负极材料来代替碳负极已经激起科学家的兴趣[5-7]。
在众多的负极材料中,非金属磷作为锂离子电池负极材料格外备受青睐。因为磷在自然界中储量丰富、成本低,且具有高达2596 mAh·g-1的理论比容量(P+3 Li++3 eLi3P)[8-9]。此外,磷具有独特的层状结构和低的原子堆积密度,有利于锂离子的迅速扩散,从而提高其循环和倍率性能。但是,磷自身导电性差,磷与锂形成Li3P的过程中会引起将近300%的体积膨胀,这会导致电极材料粉化,严重时会与集流体相脱离,因此容量衰减很快,循环性能差。为了提高磷的导电性和循环性能,通常引入非活性金属如Fe、Ni、Co等可与磷形成金属磷化物,如目前已经报道的有FeP,FeP2,CoP3,Ni2P等,非活性金属一方面可以提高电极的导电性,另一方面也可以作为缓冲矩阵,从而提高电极的循环性能[10-16]。在这些金属磷化物中,由于铁资源丰富,价格低廉,故Fe-P合金备受青睐。目前已经报道的大部分金属磷化物都是通过高能机械研磨、高温固态合成以及溶液相等方法,这些方法步骤繁琐,反应时间周期长,反应条件较苛刻,成本高,不利于规模化生产应用[10,14,17,18]。此外,这些材料在制备成电极的过程中,需要和粘结剂和导电碳混合制浆,而粘结剂一般都是非活性。粘结剂的加入使得电极整体的能量密度降低,且不利于提高电极的循环性能。与以上方法相比,电沉积是一种具有成本低、操作简单、易规模化生产的合成方法。通过调节电沉积参数如温度、pH、离子浓度、电流密度、沉积时间等,可以调控电极材料的组分、形貌和微观结构。此外,电沉积方法制备的样品可以直接用作锂离子电极,无需与导电碳和粘结剂混合[16,19,20]。因此,采用电沉积方法制备铁磷合金负极材料具有非常好的应用前景。
在本研究中,采用电沉积方法制备了非晶态Fe0.64P0.34合金,再将制备好的样品放在盐酸浸泡得到磷含量高达49%的Fe0.51P0.49合金负极。电化学研究结果表明,其初始放电容量和充电容量分别为807 mAh·g-1和545.2 mAh·g-1,首次库伦效率为67.5%,23周循环后,其可逆嵌锂容量仍然保持在445.5 mAh·g-1,为首次充电容量的82%,表现出了优异的循环性能。通过原位XRD揭示了Fe0.51P0.49与Li+的反应机理。此外,非晶态Fe0.51P0.49还是一种性能优异的软磁功能材料。本工作的研究结果为将来制备多功能合金薄膜材料提供了一种方法和理论指导,具有重要的指导意义。
1 实验
1.1 Fe0.51P0.49合金电极的制备
采用电沉积方法制备Fe-P合金。在100 mL的烧杯中,首先加入40 mL水,加热至50 ℃,然后加入硼酸,搅拌直至硼酸溶解,然后依次加入氯化铵、溴化铵、次亚磷酸钠和四水合氯化亚铁,采用稀盐酸调节溶液pH为1.5,充分搅拌均匀得到均一透明的澄清电镀液备用。电镀液中包含40 g·L-1H3BO3,20 g·L-1NH4Br,60 g·L-1NH4Cl,40 g·L-1NaH2PO2·H2O和10 g·L-1FeCl2·4H2O。采用二电极体系进行电镀。电镀时,铂片(2 cm×2 cm)用作对电极,铜箔(直径为1.2 cm的圆片)用作工作电极。电镀前,铜箔需先经过1 mol·L-1氢氧化钠(NaOH)溶液除油,蒸馏水冲洗,之后再用0.5 mol·L-1盐酸(HCl)溶液去除表面氧化物,最后用蒸馏水冲洗干净备用。电沉积时间是2 min,电流密度为44.2 A·dm-2。通过电镀制备得到的FeP合金在1 mol·L-1HCl中浸泡5 min得到非晶态的Fe0.51P0.49合金电极。最后将该非晶态的Fe0.51P0.49合金置于真空干燥箱中80 ℃下干燥10 h,备用。采用差量法(电镀后极片的质量减去电镀前极片的质量)得到电沉积制备的Fe0.51P0.49合金电极的净质量,本研究中Fe0.51P0.49合金的质量为1.0 mg。
1.2 晶体结构及形貌表征
采用粉末X射线衍射(X-ray diffraction, XRD, Philips X’pert Pro Super X-ray diffractometer, Netherlands, Cu Kα,λ=1.5418 Å)对材料进行物相分析;采用配有电子能谱(Energy-disperse X-ray Spectroscopy, EDX)附件的扫描电子显微镜(SEM, Hitachi S-4800)对材料进行微观形貌及化学成分进行分析。
1.3 电化学性能测试
在充满氩气的手套箱中(水分含量和氧含量均低于0.01×10-6)自下而上组装R2025型扣式电池,如图1所示。锂片(直径15.4 mm,厚度为1.9 mm)用作对电极。电解液为1 mol·L-1LiPF6/DMC和1 mol·L-1LiPF6/DMC+2%VC (vol)。隔膜为Celgard 2400,电池组装好后,用MSK-110型(深圳科晶有限公司)将电池加压密封,然后静置10 h以获得稳定的开路电位后进行电化学实验测试。采用LAND-CT2001D电池测试系统进行恒流充放电测试;电流密度为100 mA·g-1,电压范围是0.02~1.5 V。循环伏安(CV)测试范围是0.02~1.5 V,扫速是0.05 mV·s-1。CV测试和电镀均在Iviumstat电化学工作站上进行。
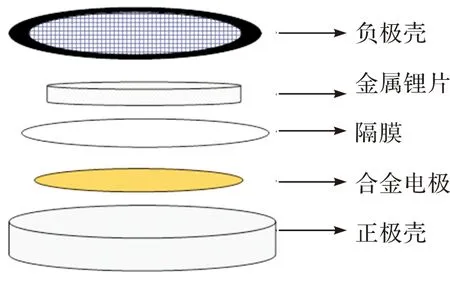
图1 扣式电池安装示意图Figure 1 Schematic diagram of button battery installation
1.4 磁性能测试
用Lakeshore 7407振动样品磁强计(Vibrating Sample Magnetometer,VSM)测量薄膜的磁性,磁场方向取平行于膜面的方向。
2 结果与讨论
2.1 结构与形貌
图2为Fe-P合金在1 mol·L-1HCl浸泡前后的XRD图。从图中可以看出,Fe-P合金在浸泡前后,除了基底Cu峰(43.1°,50.4°和74.1°)外,没有其他明显的衍射峰,在40°~50°区间均~可见一包峰,说明Fe0.64P0.34和Fe0.51P0.49合金都为长程无序、短程有序的非晶态结构。
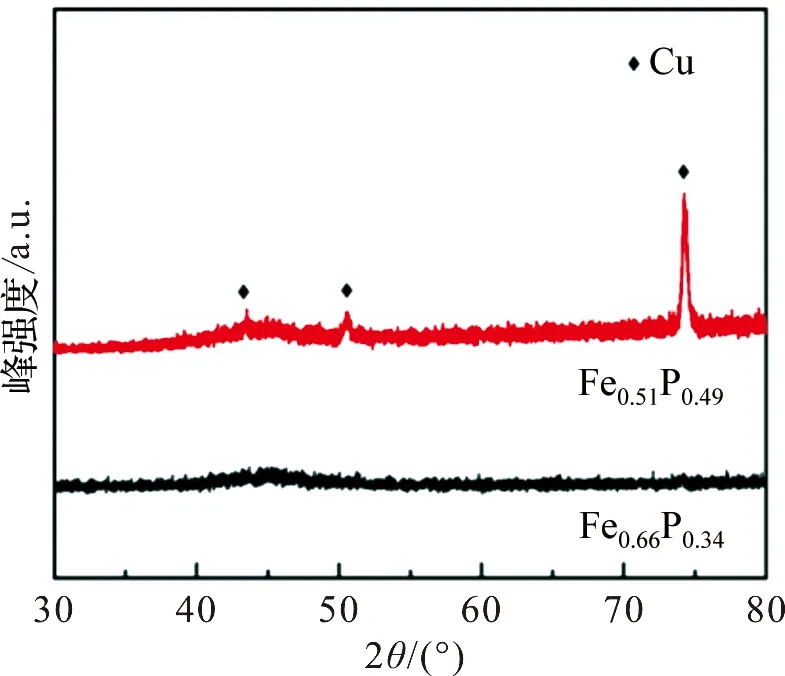
图2 Fe0.64P0.34和Fe0.51P0.49的XRD图Figure 2 XRD patterns ofFe0.64P0.34 and Fe0.51P0.49
图3是Fe-P合金电极浸泡在1 mol·L-1HCl前、后不同放大倍数(5 K, 20 K)的SEM图。从图3(a)可以看出,电镀得到的Fe0.66P0.34合金呈现类似球状的结构,球的尺寸大小不均一,在1 mm和5 mm之间,球表面非常光滑,同时可见球与球之间留有明显的空隙。从放大图3(b)可以看出,球与球之间的缝隙宽度在0.2~0.5 mm之间,棱角分明。缝隙有利于电解液渗入到电极材料内部与电解液充分接触,从而使电极材料与Li反应完全。图3(c)、3(d)为Fe-P合金电极在1 mol·L-1HCl浸泡10 min后即Fe0.51P0.49的SEM图。刚浸泡时,Fe-P合金电极表面产生大量气泡,这是由于盐酸与Fe-P合金中的单质铁反应产生氢气。当浸泡时长为10 min时,电极表面不再有气泡,说明合金中能与盐酸反应的铁单质已被反应完全,但是镀层未见脱落,说明电极与铜集流体表面吸附力强。从图3(c)、3(d)可以看出,浸泡过后的Fe0.51P0.49合金电极的表面仍然光滑,未见被腐蚀的迹象,说明非晶态Fe0.51P0.49合金具有较强的耐腐蚀性能。与酸浸泡前的Fe0.66P0.34合金表面形貌相比图3(c)、3(d),酸浸泡过后的Fe0.51P0.49合金依然为球状结构,但球与球之间的缝隙变宽,最大的可达2 μm左右。缝隙不仅有利于缩短锂离子在电极材料中的传输和扩散路径,使电极材料与电解液充分接触,从而提高电极材料的利用率;而且也能缓冲电极在充放电过程中的体积膨胀,从而提高合金电极材料的循环性能。镀层的厚度可以通过控制电沉积时间来控制。当电沉积时间为2 min时,镀层厚度为1.8 μm,可以当电极材料使用;当电沉积时间为5 min时,镀层厚度为4 μm,不宜做电极材料使用,因为镀层太厚,电解液很难进入电极内部与电极材料接触,使电极材料与Li反应不全,导致电极材料的比容量降低。因此,在本研究中,用于制备扣式电池所用的Fe-P合金电极的沉积时间均为2 min。

图3 Fe0.64P0.34和Fe0.51P0.49的SEM图Figure 3 SEM images ofFe0.64P0.34 and Fe0.51P0.49
表1为Fe0.64P0.34和Fe0.51P0.49的浸泡前后的元素含量。图4为Fe0.51P0.49的EDX图。为了进一步考察盐酸浸泡对Fe-P合金中组分的影响,我们利用EDX对盐酸浸泡前和后的Fe-P合金中Fe、P元素进行了检测,结果如表1和图4。从表2可以看出,浸泡前的Fe-P合金中Fe、P的原子百分比分别为65.6%和34.4%。而浸泡后的Fe-P合金中Fe、P的原子百分比为49%和51%。说明通过盐酸浸泡的方法,可以有效提高Fe-P合金中活性P的相对含量,降低非活性Fe的比例,从而得到高比容量的Fe0.51P0.49合金电极。基于P与Li反应生成Li3P,其理论容量高达2 596 mAh·g-1,通过计算可得,浸泡后的Fe0.51P0.49电极的理论比容量(2 596×0.348=903 mAh·g-1)远大于Fe0.66P0.34的理论容量(2 596×0.226=586.7 mAh·g-1)。图4的EDX图显示,除Fe和P两种元素外,未能检测到其他元素的能谱峰,说明非晶态Fe0.51P0.49合金稳定,表面干净,未被氧化。
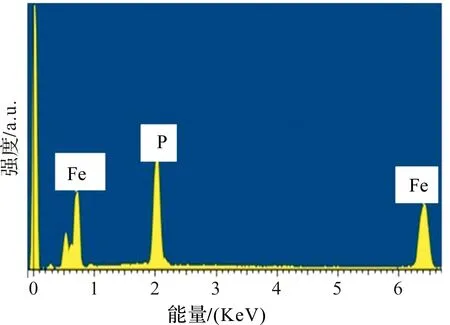
图4 Fe0.51P0.49的EDX图Figure 4 EDX of Fe0.51P0.49
Table 1 Element contents of Fe0.64P0.34和Fe0.51P0.49
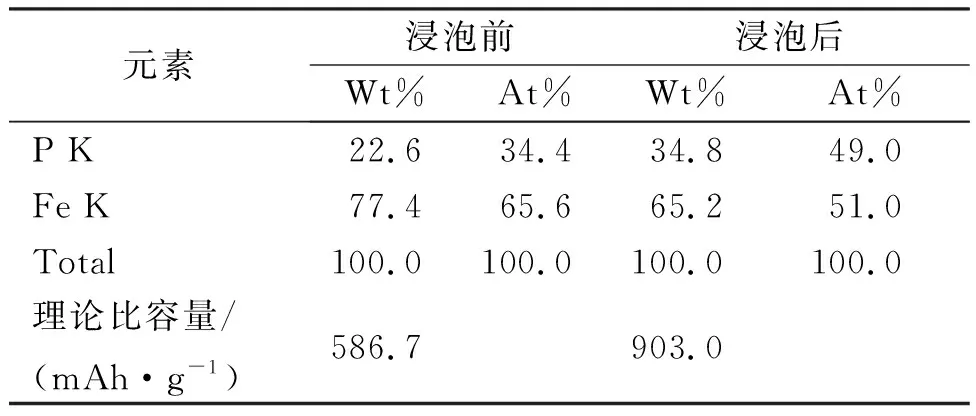
元素浸泡前浸泡后Wt%At%Wt%At%P K22.634.434.849.0Fe K77.465.665.251.0Total100.0100.0100.0100.0理论比容量/(mAh·g-1)586.7903.0
2.2 Fe0.51P0.49合金的电化学性能
电解液添加剂对VC的影响比较大。为此,考察了Fe0.51P0.49合金分别在1 mol·L-1LiPF6/DMC和1 mol·L-1LiPF6/DMC+2%VC中的电化学性能。图5为Fe0.51P0.49在不同电解液中的循环性能对比图,电压范围0.0~1.5 V,充放电电流密度均为50 mA·g-1。从图5可知,在1 mol·L-1LiPF6/DMC+2%VC电解液中,非晶态Fe-P合金的首次放电容量为807 mAh·g-1,略低于其理论容量,说明有少部分材料为能完全参与反应。首次充电容量为545.2 mAh·g-1,首次库仑效率为67.5%,23次循环后其可逆充电容量保持着445.5 mAh·g-1,为首次充电容量的82%,性能优异。而在1 mol·L-1LiPF6/DMC中,非晶态Fe-P合金的首次放电容量为794.7 mAh·g-1,充电容量为577.2 mAh·g-1,首次库仑效率为72.6%,但之后其容量衰减快,23周循环后,可逆充电容量只有234 mAh·g-1,为首次充电容量的40%。对比可知,非晶态Fe0.51P0.49合金在1 mol·L-1LiPF6/DMC+2%VC的循环性能明显优于非晶态Fe0.51P0.49合金在1 mol·L-1LiPF6/DMC中的循环性能。这主要是由于链状碳酸酯DMC在电极表面一般不能形成有效的SEI膜。而电解液添加剂VC可以改善电极SEI膜的形成电位和组成[21-23]。它会先于电解液溶剂在电极表面聚合,形成一层非常薄,且均匀致密的有机膜,有利于提高了电极的循环稳定性。
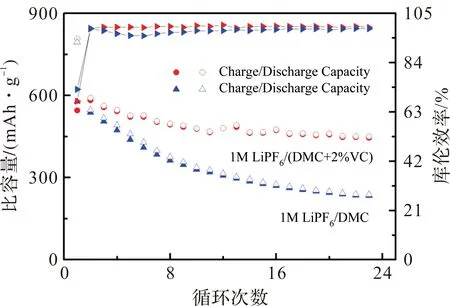
图5 Fe0.51P0.49合金分别在1 mol·L-1 LiPF6/DMC和1 mol·L-1 LiPF6/DMC+2%VC的循环性能图。Figure 5 Cycling performance of Fe0.51P0.49 alloy in 1 mol·L-1 LiPF6/DMC and 1 mol·L-1 LiPF6/DMC+2%VC
图6(a)~6(b)为非晶态Fe0.51P0.49合金在1 mol·L-1LiPF6/DMC和1 mol·L-1LiPF6/DMC+2%VC的充放电曲线,图6(c)~6(d)为微分容量曲线图。由图6(a)的充放电曲线可知,首次放电过程中,从开路电位到0.8 V时,曲线上出现一个很大的斜坡,而在第2周之后消失,这主要是由于电解液的分解、电极表面形成SEI膜及极化等原因,该部分的容量为不可逆容量。随后缓慢下降至0.2 V时出现一个很长的电压平台,主要是由于Li+与非晶态Fe0.51P0.49合金发生合金化反应产生Li3P与纳米Fe[18]。在充电曲线中,在0.0~1.0 V出现一长斜坡,主要是发生Li3P的去合金化反应。图6(c)为其微分容量曲线,进一步说明Fe0.51P0.49与Li的合金化反应电位。从图6可知,在1.7 V出现一明显的氧化峰,这对应于电解液的分解或SEI膜的生成等,在0.03 V出现很强的电流峰,对应于Li+与非晶态Fe0.51P0.49合金发生合金化反应产生Li3P。第2周之后,在0.63 V出现了新的氧化峰,同时发生合金化反应的电位由第一次循环的0.02 V正移至0.15 V,说明Li+与P反应可能要有多个反应过程,并不是一步形成Li3P。这在之后利用原位XRD研究其充放电机理中得到了证实。还原过程中,0.9 V对应于Li3P的去合金化反应。与非晶态Fe0.51P0.49合金在1 mol·L-1LiPF6/DMC的充放电曲线和微分容量曲线相比,可以发现其电位平台和峰电位基本和加有VC的一致,但微分容量曲线中峰电位的强度减弱的很快。结果说明电解液添加剂VC不会影响非晶态Fe0.51P0.49合金电极材料与Li+的反应机理,但有助于形成稳定的SEI膜[24],缓解容量衰减,从而提高循环性能。这与前面的分析结果一致。

图6 非晶态Fe0.51P0.49合金分别在1M LiPF6/DMC和1M LiPF6/DMC+2%VC的充放电曲线及微分容量曲线图Figure 6 Differential capacity curves of amorphous Fe0.51P0.49 alloy in 1M LiPF6/DMC and1M LiPF6/DMC+2%VC
为了进一步研究非晶态Fe0.51P0.49合金电极的与锂的反应机理。我们采用原位XRD研究了非晶态Fe0.51P0.49合金的首次充放电过程。图7为非晶态Fe0.51P0.49合金电极放电至不同电位的原位XRD图。图8为非晶态Fe0.51P0.49合金电极充电至不同电位的原位XRD图。图7(a)为非晶态Fe0.51P0.49合金电极在开路电位(3.2 V)下的XRD图。本实验采用Cu为集流体,其衍射峰较强,因此,基底Cu的四个明显特征峰(43.0°、50.4°、74.0°和89.9°)均被检测到。除Cu的特征峰外,谱图中在35°~40°范围只有一个宽峰,说明Fe0.51P0.49为非晶态,与前面的分析结果一致。在图7(b)中,当放电至1.5 V时,仍然可见35°~40°的包峰,但衍射峰强度较弱。在图7(c)中,当放电至0.8 V时,在45°出现新的衍射峰,这对应于Fe,说明非晶态Fe0.51P0.49与Li开始反应,产生Fe。在图7(d)中,当继续放电至0.65 V时,在37°和39°新增两个衍射峰,根据物相分析为LiFeP,说明Fe0.51P0.49与Li+在0.65 V开始发生合金化反应产生中间相LiFeP。在图7(e)中,当继续放电至0.4 V,图中37°和39°处对应的LiFeP衍射峰增强,同时在47.5°和55°又出现了两个新增衍射峰,对应于Li3P相。在图7(f)中,当再继续放电至0.02 V,37°和39°所对应的LiFeP衍射峰依然存在,同时出现Li3P相(34.5°、47.5°和55°)。这说明Fe0.51P0.49与Li+反应先生成LiFeP中间相,之后再转变成Li3P相。在充电过程中,当充电至0.95 V时,图8(a)显示LiFeP仍然存在,但Li3P相的特征峰完全消失。当继续充电至1.5 V时如图8(b),只观察到Fe相,除Cu的峰外,未见其他明显衍射峰。说明Li3P相在充电过程中,完全发生去合金化反应,可逆性好。

图7 非晶态Fe0.51P0.49合金电极放电至不同电位的原位XRD图(未标识的衍射峰均为Cu)Figure 7 In situ XRD patterns of amorphous Fe0.51P0.49 alloy after discharged to different potentials (The diffraction peaks not identified in patterns are ascribed to Cu substrate)
综合以上的实验结果,通过原位XRD检测到非晶态Fe0.51P0.49合金电极在充放电过程中的相变,即放电至0.65 V时,非晶态Fe0.51P0.49与Li+发生合金化反应产生LiFeP中间相,当继续放电至0.02 V时,则出现Li3P,因此,非晶态Fe0.51P0.49合金电极在充放电过程中经历了两个相变过程,其反应表达式为:Fe-P+3 Li++3 e→LiFeP→Fe+Li3P。
该分析结果与前面微分容量曲线的分析结果一致。

图8 非晶态Fe0.51P0.49合金电极充电至不同电位的原位XRD图(未标识的衍射峰均为Cu)Figure 8 In situ XRD patterns of amorphous Fe0.51P0.49 alloy after charged to different potentials. (The diffraction peaks not identified in patterns are ascribed to Cu substrate.)
2.3 Fe-P合金的磁性能(VSM)表征
Fe-P合金除能作为电池材料之外,它还是一种很好的磁性功能材料。图9为Fe0.66P0.34与Fe0.51P0.49合金的磁滞回线。利用振动样品磁强计对两种不同组分Fe0.66P0.34和Fe0.51P0.49合金的磁性能进行测量,磁场方向平行于样品表面的方向,测试结果如图9。从图9中可知,Fe0.66P0.34和Fe0.51P0.49的磁滞回线均细小而窄,矫顽力不大,表现出较好的软磁性能。与Fe0.66P0.34相比,Fe0.51P0.49的剩磁Br,饱和磁感应强度变小,而矫顽力则变大。Fe0.66P0.34的矫顽力为54 Oe(4 266 A·m-1),而Fe0.51P0.49的矫顽力为115 Oe(9 085 A·m-1)。这说明合金完全处于非晶态时,组分中P含量相对比例增大,合金的矫顽力增大。这是由于矫顽力是磁损耗的控制因子,在非晶材料中存在对总矫顽力有贡献的五种钉扎效应中,对于用电沉积法制备的非晶合金,其中化学短程序成团区Hc(SO)、局域结构重排的驰豫效应Hc(rel)和由于缺陷微结构造成的体钉扎Hc(s)起了主要的作用,Fe0.66P0.34中单质Fe被溶解得到Fe0.51P0.49合金,由于P与Fe具有较强的化学亲和力,形成以P为中心周围包围着Fe原子的结构,且P含量越大,合金的无序度增加,引起了Hc(SO)和Hc(rel)的增加,且随磷含量的增加,非晶合金中存在的以自由体积为特征同时还包含少量较大空洞的结构缺陷也增多,结构缺陷对畴壁可逆位移的阻滞作用将增大,从而引起Hc的增加。这就造成了Fe0.51P0.49的总矫顽力Hc比Fe0.66P0.34的矫顽力大。

图9 Fe0.66P0.34与Fe0.51P0.49合金的磁滞回线Figure 9 Hysteresis loops of Fe0.66P0.34and Fe0.51P0.49 alloys
3 结 论
本文通过一步电沉积和酸浸泡相结合的方法制备了一种高磷非晶态Fe0.51P0.49合金材料并研究了其电化学性能与磁性能。电化学研究结果表明,电解液VC有助于形成稳定的SEI膜,有助于提高Fe0.51P0.49合金的电化学性能。其初始放电容量和充电容量分别为807 mAh·g-1和545.2 mAh·g-1,首次库伦效率为67.5%,23周循环后,其可逆充电容量为445.5 mAh·g-1,为首次充电容量的82%,表现出了优异的循环性能。通过原位XRD揭示了Fe0.51P0.49与Li+的反应机理。磁性能结果表明,Fe0.51P0.49的矫顽力曲线呈典型的“S”型曲线,矫顽力为115 Oe(9 085 A/m),表现为软磁性能。因此,通过电沉积方法制备的非晶态Fe0.51P0.49合金是一种很好的多功能材料。
猜你喜欢
铝加工(2022年3期)2022-11-24
电池(2022年4期)2022-11-07
材料与冶金学报(2022年2期)2022-08-10
新能源汽车供能技术(2021年1期)2021-10-14
粉末冶金技术(2021年3期)2021-07-28
储能科学与技术(2020年2期)2020-04-04
电子制作(2019年23期)2019-02-23
中国校外教育(中旬)(2018年9期)2018-09-30
汽车实用技术(2015年8期)2015-12-26
燕山大学学报(2015年4期)2015-12-25
