
ZIF67衍生硫化钴/多孔碳复合催化剂的制备及其电催化性能
2019-08-08 06:52李京修薛建军何娉婷
无机化学学报 2019年8期
李京修 赵 媛,2 薛建军 何娉婷 王 玲*,
(1南京航空航天大学材料科学与技术学院,南京 210016)
(2金陵科技学院材料工程学院,南京 211169)
0 引 言
随着社会的不断发展,人类日益增长的能源需求导致了化石燃料的迅速消耗,与此同时化石燃料的燃烧带来的污染导致了严重的全球性环境问题[1]。为了缓解人类对自然能源的严重依赖,对清洁可再生的能源技术的探索显得尤为迫切,燃料电池作为一种将化学能转换为电能的能源转换装置,具有能量密度高、二次污染少的优点,成为了新一代绿色可再生能源中的佼佼者,然而燃料电池中缓慢的阴极氧还原反应(ORR)动力学严重制约了燃料电池的商业化进程[2]。传统的Pt基催化剂虽然具有较低的过电位和较高的ORR活性,但是由于其地壳存储量低、价格高昂,容易被毒化的缺点严重制约了其商业化的进程。因此开发价格低廉且高效的ORR催化剂成为了科研工作者重要的研究方向[3-5]。
金属有机骨架(Metal organic frameworks,MOFs)材料是一种由金属离子和多齿有机配体通过配位键构成的具有三维多孔结构的有机-无机复合材料,近年来得到广泛的关注。与传统的多孔材料相比,MOFs具有结构多样、易于功能化修饰、孔径可调控等优点[6-7],目前在气体的吸附与分离、催化、药物输送、传感等领域得到了广泛的应用[8-9]。最近许多的研究者将研究的重点放在以MOFs材料为自牺牲前驱体或模板原位合成碳载金属、金属氧化物、金属碳化物,该类型的碳载催化剂在合适的条件下不仅可以保留MOFs本来的形貌,还可以有效地增强催化剂与碳载体之间的作用[10-12]。2011年Ma等[13]以Co基的咪唑酯为前驱体通过直接热解制备出Co掺杂多孔碳复合催化剂,并在酸性条件下研究了复合催化剂的ORR性能。研究结果显示,750℃热处理后复合催化剂在0.1 mol·L-1HClO4中起始点位达到0.83 V,半波电位达到 0.68 V,根据 Koutechy-Levich(K-L)方程计算得到ORR过程的转移电子数集中在3.2~3.5,这对于催化剂的ORR性能是非常有利的。南洋理工大学的楼雄文课题组[14]创新性地以ZIF67为反应前驱体,以硫代乙酰胺为S源,通过精确控制反应的时间制备出一系列中空结构的纳米笼,热处理后成功制备出双核壳的Co-C@Co9S8复合催化剂,其独特的催化剂结构使得该催化剂在碱性条件下表现出优异的ORR性能、稳定性能和抗甲醇性能。MOFs材料虽然在电催化方向展现出良好的应用前景,但MOFs材料较差的导电性和其应用在水溶液中时的不稳定性导致了其电化学应用性能不佳。近年来,人们采用多种方法以期获得导电性与稳定性更优异的MOFs及其衍生材料,如:将MOFs与具有高导电能力的高分子聚合物、石墨烯、碳纳米管等进行复合;对MOFs表面进行化学修饰;使用具有多个苯环平面共轭结构的配体;直接高温热解转化为相应碳基材料等[15-16]。MOFs衍生的复合催化剂多数需要在高温条件下热解来达到造孔的目的以增加催化剂的比表面积,然而在高温碳化过程中容易使催化剂团聚成大颗粒,且碳化过程中难以控制合成的活性位点,因此如何使该类催化剂具有大的比表面积和更多的活性位点成为研究的重点。
以ZIF67为前驱体,硫代乙酰胺为硫源,采用离子交换与热处理相结合的方法制备出高分散性且大比表面积的硫化钴/多孔碳(CoS/C)复合催化材料,并研究了硫化时间对复合催化剂形貌结构及ORR性能的影响。研究结果显示,随着硫化时间的增加,硫化钴颗粒逐渐增大,且催化剂中碳材料的无序程度出现先减小后增大的趋势,在反应时间为10 min时,复合催化剂在0.1 mol·L-1KOH中表现出良好的电催化活性。
1 实验部分
1.1 材料制备
催化材料的制备:称取2.91 g六水合硝酸钴(Co(NO3)2·6H2O)加入到 125 mL 的甲醇溶液中,配成溶液A。称取3.28 g的2-甲基咪唑加入到125 mL的甲醇溶液中,搅拌溶解配成溶液B。将溶液B迅速加入到溶液A中,室温条件下反应24 h,得到蓝紫色混合溶液。将反应后的溶液抽滤,用甲醇溶液清洗3~5次,冷冻干燥后备用。
称取上述前驱体0.2 g,分散到20 mL乙醇溶液中,向分散液中加入10 mL含有0.2 g硫代乙酰胺的乙醇溶液,90℃条件下反应10 min,抽滤清洗干燥后,氮气氛围中将混合物在650℃下热处理2 h,升温速率为10℃·min-1,最终产物即为硫化钴/多孔碳(CoS/C)复合催化剂。
1.2 材料结构表征
催化剂的颗粒形貌采用日本HITACHI SU-4800型场发射扫描电镜(SEM,操作电压为1 kV)以及捷克Tecnai G2 T20透射电子显微镜进行表征(TEM,加速电压为200 kV);催化剂的晶体结构采用日本理学UltimaⅣX射线衍射仪进行表征,仪器的射线源为 Cu Kα,λ=0.154 056 nm,管电流为 100 mA,管电压为40 kV,Ni为滤波片,扫描角度范围为10°~80°,扫描角度分辨率为 0.02°,扫描速度为 2°·min-1。采用美国Micromeritics公司ASAP 2010型氮气吸附仪并通过使用BET方法及BJH模型计算得出催化剂的比表面积和孔径分布。采用英国Renishaw公司生产的Renishaw inVia显微拉曼光谱仪进行表征。 测量范围:200~3 500 cm-1,激光器采用 532 nm的激光光源。测试中采用Thermo ESCALAB 250型X射线光电子能谱仪,Al Kα为光源,以C1s(284.4 eV)峰来校准其它元素的XPS峰,用来分析催化材料表面元素的价态和存在形式。
1.3 电化学性能测试
工作电极的制备是将5 mg催化剂分散到含有950μL的乙醇和水的混合溶液中 (体积比为4∶1)后加入50μL 5%(w/w)的 Nafion溶液,超声分散 30 min制备成均匀墨汁溶液。滴制电极前,在麂皮绒布上用0.5μm的抛光粉(Al2O3)对直径为5 mm的玻碳电极进行充分研磨。研磨后,超声清洗干燥后备用。用微量进样器将10μL的墨汁电解液滴在玻碳电极的表面,室温下干燥后即制得工作电极。
催化剂的ORR电催化性能评价在上海辰华CHI660D电化学工作站与美国Pine型旋转圆盘电极装置(RDE)上进行,采用三电极体系进行测试。三电极分别以附载催化剂的玻碳电极作为工作电极,导电碳作为对电极,Hg/HgO电极作为参比电极。在O2或 N2饱和的 0.1 mol·L-1的 KOH溶液中进行测试。
2 结果与讨论
2.1 ZIF67的形貌表征
对制备的ZIF67进行SEM和TEM表征(图1)。实验制备的ZIF67为粒径均一的正十二面体。由图1(c)的粒径分布图可以得知,ZIF67的平均粒径在80 nm左右,均一的粒径为CoS/C复合催化剂的制备提供了良好的基础。
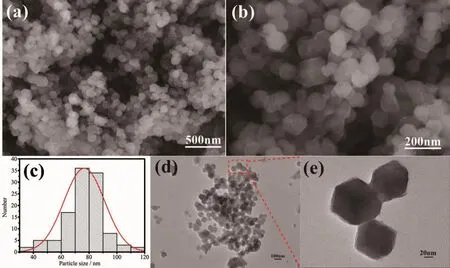
图 1 ZIF67 的 SEM 图 (a,b)、粒径分布图 (c)和 TEM 图 (d,e)Fig.1 SEM images (a,b),particle size distribution diagram (c)and TEM images (d,e)of ZIF67
2.2 CoS/C复合催化剂的物理表征
硫代乙酰胺与ZIF67的反应时间会影响CoS/C复合材料的电化学性能,图 2(a~f)分别反应时间为0、5、10、15、20、30 min 时 CoS/C 的 TEM 图。 反应 5 min时,热处理后复合催化剂保持ZIF67的原始形貌,在0~10 min内随着反应时间的增加,硫化钴的晶粒尺寸逐渐增加,此时催化剂具有良好的分散性,而在15~30 min时,复合催化剂出现明显的团聚,这对于催化剂自身的导电性极为不利,因此合理控制硫代乙酰胺与MOF材料的反应时间是非常重要的。

图2 不同硫化时间制备的CoS/C的TEM图Fig.2 TEM images of CoS/C prepared at different vulcanization times

图3 硫化时间为10 min时制备的CoS/C的XRD图 (a)和不同硫化时间下制备的CoS/C的Raman图谱 (b)Fig.3 XRD pattern of CoS/C obtained at vulcanization time of 10 min (a)and Raman spectra of CoS/C obtained with different vulcanization times
图 3(a)为 CoS/C 的 XRD 图,图中在 30.6°、35.3°、46.9°、54.4°出现的特征峰分别对应六方晶系CoS(PDF No.65-3418)的(100)、(101)、(102)、(110)晶面[17],说明复合催化剂中的主要成分为CoS。为了进一步探究反应时间对复合催化剂结构的影响,对不同反应时间制备的CoS/C进行了拉曼测试,如图3(b)所示,位于1 000 cm-1以下的特征峰归属于无机物,其中在 474.6、526.71、679 cm-1出现了 3个 CoS的特征峰[18-19],且随着硫化时间的增加,3个特征峰基本呈明显的趋势,这意味着CoS在催化剂中的含量随着硫化时间的增加而不断增多。采用Raman光谱分析碳材料时,常常出现2个特征峰,分别为D峰(1 350 cm-1附近)和 G 峰(15 80 cm-1附近)。 其中 D峰是由边缘不饱和碳原子(sp3键)引起的,G峰是由sp2碳原子的面内振动引起的,可用来表征碳材料的石墨结构中sp2杂化键结构的完整程度。通常用D峰与G峰的强度比值(ID/IG)来衡量物质不规则度和sp2域的平均尺寸,以判断物质有序性以及碳材料自身的缺陷程度与缺陷密度[20-21]。
通过计算,可以得到不同硫化时间下CoS/C的ID/IG数值(表1)。硫化时间为0和5 min时,由于S2-未大量进入MOF结构中,热解后复合催化剂保持了本身的MOF结构,这在一定程度上影响了复合催化剂的碳化程度。在硫化时间为10 min时,ID/IG值最小。随着硫化时间的增长,S2-进入MOFs结构的量不断增多。由于S元素容易升华,在热处理过程中会有大量的S挥发,增加了复合催化剂的孔隙率,这就使得在硫化时间为15~30 min的过程中,催化剂的ID/IG值逐渐增大,催化剂中碳材料的无序度增加。
硫化10 min得到的CoS/C复合催化剂的比表面积和孔径分布通过N2吸附-解吸等温线测定。如图4(a)所示,复合催化剂的N2吸附-脱附等温线呈LangmuirⅣ型等温线,在较低相对压力时,氮气吸附急剧增加,说明制备得到的催化剂中存在微孔。相对压力继续增加时,氮气吸附增加趋势放缓。同时复合催化剂在相对压力P/P0>0.9时都存在滞后环,说明材料是介孔材料。计算可知复合催化剂的BET表面积为 133 m2·g-1,主要孔径为约为 5.759 nm(图 4(b)),累积孔体积为0.148 5 cm3·g-1。这种多孔结构可以提供更多活性位点以增强电催化剂的ORR性能[16,22]。
XPS测试可以进一步对多孔碳硫化钴复合材料中元素的存在形式进行分析。XPS全谱图(图5(a))中出现明显的 C1s(~284.4 eV)、Co2p(~780 eV)、O1s(~532 eV)、S2p(160~170 eV)和 N1s(~400 eV)特征峰,进一步证明了CoS/C复合催化剂的元素组成。对每个元素的谱图进一步分析可以准确得出同一元素不同的存在形式。复合材料中的N主要来源于MOF结构中的有机配体,因此材料中N的含量比较低。经过高温热解之后,复合材料中的N主要以石墨氮(401.7 eV)、吡啶氮(398.6 eV)和吡咯氮(399.8 eV)的形式存在(图 5(b))[23]。 图 5(c)为 Co2p 的高分辨图谱,在778.4 eV处的峰对应CoS/C的Co2p3/2结合能,793.7 eV处的峰对应Co2p1/2结合能,与此同时,伴随着802.4和785.9 eV处震激卫星峰的出现,证明了 Co 主要以 Co2+和 Co3+两种形式存在[24]。 图 5(d)表明复合材料中S元素主要存在3个特征峰,其中168.6 eV处特征峰归属于SO42-,这主要是由于加入的硫源在空气中氧化形成的,而位于162.9 eV的S2p1/2和161.5 eV的S2p2/3的特征峰是因Co-S键的存在产生的[25]。

表1 不同硫化时间(t)制备的CoS/C的I D/I GTable 1 I D/I G of CoS/C obtained w ith different vulcanization times (t)

图4 硫化10 min得到的CoS/C的氮气吸附-脱附曲线 (a)和孔径分布图 (b)Fig.4 N2 adsorption-desorption isotherm (a)and pore size distribution profile (b)of CoS/C obtained with vulcanization time of 10 min

图5 硫化10 min得到的CoS/C的XPS图谱Fig.5 XPS spectra of CoS/C obtained with vulcanization time of 10 min
2.3 电化学性能测试
图6为不同硫化时间得到的复合催化剂在N2和O2饱和的0.1 mol·L-1KOH中测试的循环伏安(CV)曲线,扫描范围为 0.2~1.1 V(vs RHE),扫描速率为20 mV·s-1。由图可知,CoS/C催化剂在N2气氛下没有明显的峰出现,说明催化剂在N2氛下稳定存在,而在O2条件下均出现1个明显的氧还原峰,说明在O2氛围下催化剂会催化氧气的还原,具有一定的ORR活性。ORR是一个先吸附后还原的过程,因此CV曲线中氧还原电位越正,复合催化剂的ORR性能越好[26]。硫化时间从0到30 min时,氧还原电位依次是 0.715 9、0.721 5、0.770 9、0.734 8、0.733 5和0.675 6 V(vs RHE),说明随着硫化时间的增加,氧还原电位先增加后减小。当硫化时间为10 min时,氧还原电位最正,此时催化剂具有最佳的ORR性能,这与TEM与Raman的分析结果一致。
对不同硫化时间制备的CoS/C复合催化剂在O2饱和的0.1 mol·L-1KOH中测试ORR性能,扫描范围为 1.0~0.2 V(vs RHE),扫描速率为 5 mV·s-1,转速为1 600 r·min-1,测试结果如图7所示。由图可知,随着硫化时间的增加,复合催化剂的起始电位和半波电位先增加后减小。当硫化时间10 min时,有最大的起始电位、半波电位值,说明其拥有最佳的ORR性能。对比10 min制备的CoS/C复合催化剂与商业20%Pt/C催化剂在1 600 r·min-1时的LSV曲线可知,CoS/C复合催化剂的半波电位(E1/2)仅比商业Pt/C催化剂低31 mV,说明硫CoS/C复合催化剂具有良好的ORR性能。
进一步的对复合催化剂的ORR机理进行分析。图8(a)是硫化10 min得到的复合催化剂在不同转速条件下得到的LSV曲线,在高电位时不同转速下的极限电流密度值区分不大,甚至有所重合。随着电位值的降低,极限电流密度值开始互相偏移,产生区分。在低电位时,极限电流密度值呈现等差分布,转速越高则极限电流密度值越高。根据不同转速下的LSV曲线拟合的K-L方程的结果计算不同电位下ORR 过程中转移电子数 n(图 8(b)),可知在不同电位下复合催化剂表面ORR过程的n的平均值为3.72,接近于4,说明了该反应符合理论中的ORR四电子过程。
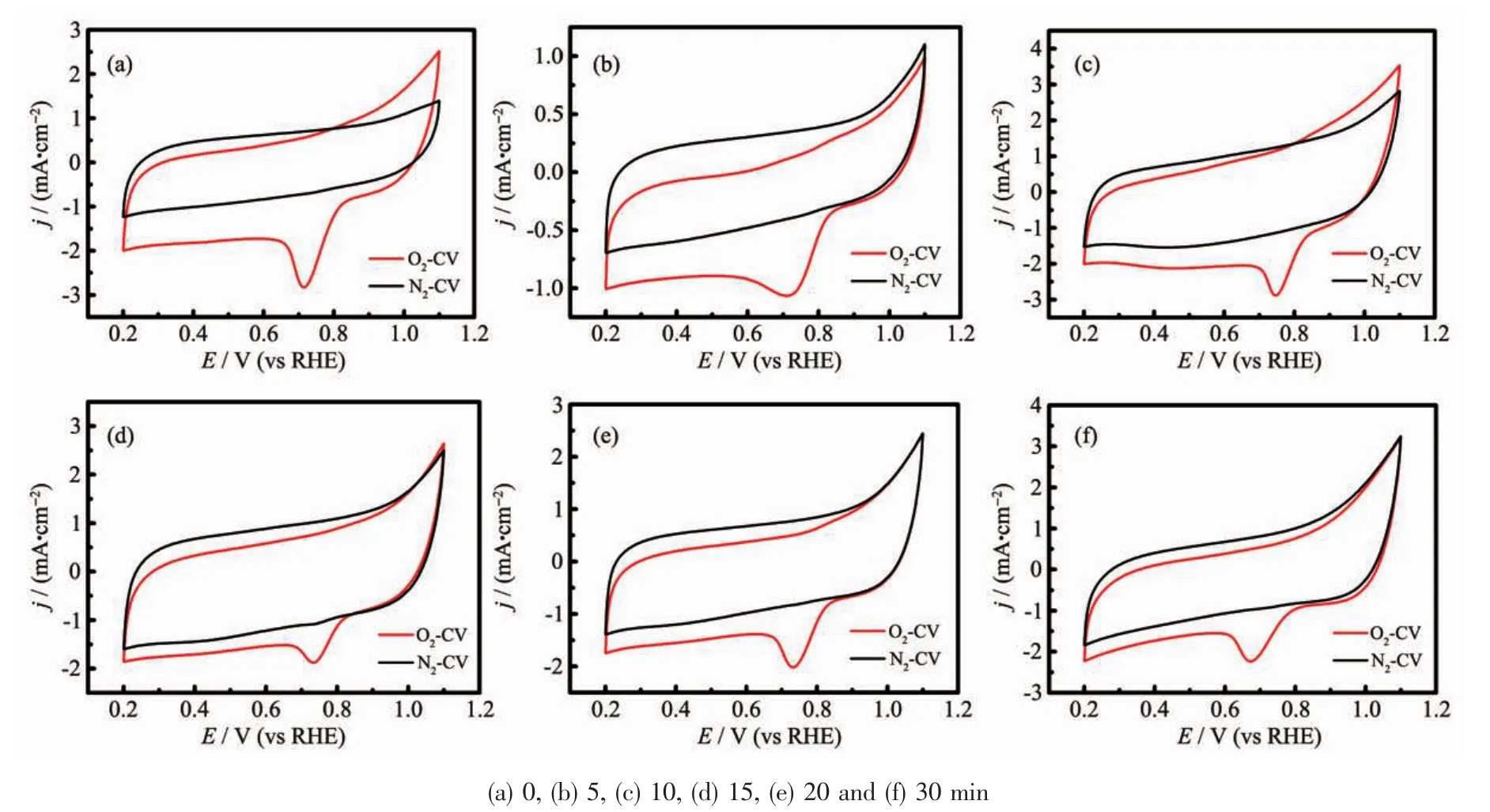
图6 不同硫化时间制备的CoS/C的CV曲线Fig.6 CV curves of CoS/C obtained with different vulcanization times

图7 不同硫化时间制备的CoS/C的LSV曲线Fig.7 LSV curves of CoS/C obtained with different vulcanization times

图8 硫化时间为10 min时复合催化剂在不同转速下LSV曲线 (a)和相应的K-L曲线 (b)Fig.8 LSV curves under different rotation rates of composite catalyst obtained with vulcanization time of 10 min (a)and corresponding K-L plots (b)
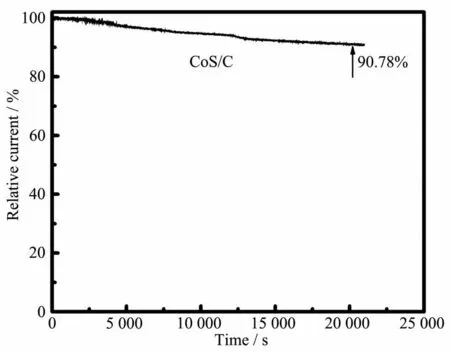
图9 硫化时间为10 min得到的CoS/C复合催化剂的稳定性能Fig.9 Cycle stability of CoS/C composite catalystobtained with vulcanization time of 10 min
催化剂的稳定性也是评价催化剂的重要指标,实验中采用计时电流法测定催化剂的稳定性能。在O2饱和的 0.1 mol·L-1KOH 中,在 0.7 V(vs RHE)的电压条件下监测复合催化剂表面的电流随时间的变化(图9)。经过20 000 s后,复合催化剂的电流衰减至原始电流的90.78%,表明复合催化剂的具有良好的稳定性能。
3 结 论
以ZIF67为前驱体,硫代乙酰胺为硫源,通过离子交换法与热处理相结合制备出CoS/C复合催化材料,在碱性条件下该复合催化剂具有与商业20%Pt/C催化剂相媲美的ORR活性,其半波电位仅比Pt/C催化剂低31 mV。研究结果显示,随着硫化时间的增加,硫化钴颗粒逐渐增大,且催化剂中碳材料的无序程度出现先减小后增大的趋势。在硫化时间为10 min时,复合催化剂在0.1 mol·L-1KOH中表现出良好的电催化活性,且ORR过程中的平均转移电子数可达到3.72,接近于4,说明O2在该催化剂表面发生的是四电子转移过程,这对于ORR是十分有利。同时该复合催化剂在碱性条件下也表现出良好的稳定性能。
猜你喜欢
弹性体(2022年3期)2022-11-15
轮胎工业(2022年2期)2022-07-19
汽车工程师(2021年12期)2022-01-18
中国听力语言康复科学杂志(2021年6期)2021-12-21
食品安全导刊(2021年20期)2021-11-28
陶瓷学报(2021年4期)2021-10-14
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
大经贸(2020年1期)2020-04-07
商品与质量(2019年42期)2020-01-17
电子制作(2018年9期)2018-08-04
