
瞬时受体电位通道1在大鼠脑缺血后钙超载致神经元损伤中的作用机制
2019-08-06 03:10赵永林赵君杰黄廷钦马旭东宋锦宁
山西医科大学学报 2019年7期
张 明,吴 媛,赵永林,赵君杰,黄廷钦,马旭东,宋锦宁*
(1西安交通大学第二附属医院神经外科,西安 710004;2西安交通大学第二附属医院重症医学科;3西安交通大学第二附属医院肿瘤病院;4西安交通大学第一附属医院神经外科;*通讯作者,E-mail:jinnings@126.com)
脑卒中是导致老龄人群致残和死亡的重要原因,其中约80%患者为缺血性卒中。缺血性脑损伤是影响患者远期预后的主要原因[1],一旦缺血开始形成,在数分钟至数小时内就会发生序贯性加重的脑损伤,缺血导致相关区域神经血管单元功能受到严重影响,尤其是神经元处于高代谢状态,在缺血影响下导致细胞内外能量代谢失衡,在此过程中神经元细胞更易受到损害,引起细胞信号传导异常,除引起兴奋性毒性外[1],还有可能产生因Ca2+超载导致的非兴奋性毒性机制。近年来,有研究认为Ca2+可通透性瞬时受体电位通道1(transient receptor potential channel 1,TRPC1)是非兴奋性毒性机制相关蛋白[2]。体内实验表明,敲除小鼠海马神经元的TRPC1,可明显减少动作电位诱发的兴奋性突触后电流,对突触传递产生影响[2]。进一步的研究证实[3],TRPC1缺失可能通过扰乱多种生物过程引起纹状体神经细胞凋亡,包括内质网功能、氧化应激和凋亡相关信号通路,TRPC1可能是调节小鼠纹状体细胞存活和死亡的关键参与者。但TRPC1在脑缺血后神经元中的表达变化如何,是否与脑缺血后钙超载导致的神经元凋亡相关,抑制TRPC1引起的钙内流是否对脑缺血后神经元起到保护作用,目前具体的作用机制尚不明确。因此,本研究采用四血管阻塞法建立大鼠脑缺血模型,探讨TRPC1离子通道在缺血后神经元钙超载中的作用,以揭示TRPC1通道参与脑缺血后神经元凋亡及突触损伤的机制,寻找临床治疗新靶点。
1 材料与方法
1.1 实验动物及主要试剂、仪器
6周龄雄性SD大鼠120只,SPF级,体质量220-260 g,由西安交通大学动物实验中心提供(许可证号:SCXK(陕)08-018)。SKF96365(S7809,Sigma-Aldrich,美国);二甲基亚砜溶液(DMSO,D8418,Sigma-Aldrich,美国);细胞膜蛋白提取试剂盒(Thermo Scientific,美国);NeuN(Chemicon,MAB377B,美国)TRPC1兔单克隆抗体(Abcam,美国);β-actin鼠单克隆抗体(Santa Cruz,美国);辣根过氧化物酶标记的IgG二抗(Santa Cruz,美国);PVDF膜(Millipore,美国);TUNEL凋亡试剂盒(Promega,美国);凝胶成像系统(JS-380A,中国);图像采集与分析系统(Leica-Q550CW,德国);透射式电子显微镜(H-600型,日本);脑立体定向仪(Stoelting Ultra Precise 51600,美国)。
1.2 大鼠分组处理及全脑缺血模型的建立
120只SD大鼠被随机分为脑缺血模型组(n=50),SKF96365干预模型组(n=50),DMSO干预对照模型组(n=10),正常对照组(n=10)。脑缺血模型组及SKF96365干预模型组各分为5个亚组,分别为缺血后1 d,3 d,5 d,7 d,10 d组,每组10只。
大鼠术前禁食禁饮4-6 h,10%水合氯醛腹腔注射麻醉。颈前部剃毛备皮,常规消毒,暴露双侧颈总动脉,分离并完全结扎双侧锁骨下动脉。当大鼠翻正反射将要恢复,即有翻身动作但尚不能翻身时,迅速用微动脉夹夹闭双侧颈总动脉,使大脑血供完全消失,可见大鼠挣扎数秒后于60 s内昏迷,同时可见大鼠四肢上举,痛觉反射消失,反正反射消失,双侧瞳孔散大,眼球呈灰白色,计时15 min,放开动脉夹迅速缝合伤口。大鼠置于恒温箱中进行术后恢复。SKF96365干预模型组将5 μl TRPC1阻断剂SKF96365溶于DMSO,终浓度为10 μmol/L,于建模前30 min经立体定向侧脑室微注射(前囟后1 mm,旁开1.5 mm,深3.5 mm);DMSO干预对照模型组使用等量DMSO行侧脑室注射,其他操作与全脑缺血模型组相同。正常对照组不予任何处理。
1.3 免疫形态学及电镜研究
各组大鼠在缺血后1,3,5,7,10 d分别进行形态学标本采集[4]。10%水合氯醛以0.5 ml/kg麻醉,用40 g/L的多聚甲醛灌流固定。石蜡包埋并制成5 μm切片,经烘片、脱蜡、梯度酒精脱水及抗原修复后,滴加封闭血清,在湿盒中37 ℃封闭30 min。滴加适宜浓度的一抗置湿盒中37 ℃ 1 h,再洗去一抗,滴加荧光二抗,室温湿盒中避光孵育1 h,洗二抗后滴加DAPI,之后用甘油封片,在荧光显微镜下立即观察并采集图像。采用SABC法进行组织化学染色并在光镜下进行观察。将海马标本用电镜固定液固定,采用常规电镜标本制作方法,经清洗、脱水、浸透、包埋和修块后作超薄切片,在透射电镜下观察。
1.4 海马神经元细胞内Ca2+浓度([Ca2+]i)检测
取新鲜海马剪碎后置于1.5 ml冰Hank’s液中,吹打10 min,过200目滤网。用含100 ml/L小牛血清的DMEM培养液制成单细胞悬液(106个/ml)。取细胞悬液1 ml离心,弃上清,37 ℃预温后加入7.5 μl的Fura-2AM(终浓度为6 μmol/L);另取同一细胞悬液1 ml未负载Fura-2AM作为空白对照。单细胞悬液37 ℃复温后在流式细胞仪上检测荧光强度[4,5]。激发波长为380 nm,发射波长为510 nm,分别测定F、Fmax、Fmin。F为380 nm激发波长测得的荧光强度值;Fmax为最大荧光强度值,即加入Triton X-100(终质量浓度为1 nmol/L)后所测得的荧光强度值;Fmin为最小荧光强度值,即在Fmax基础上加入EGTA(终浓度为5 nmol/L)后所测得的荧光强度值。胞内Ca2+浓度计算:[Ca2+]i=Kd(F-Fmin)/(Fmax-F),其中Kd=224 nmol/L。
1.5 Western blot检测蛋白表达
各组大鼠在缺血后1,3,5,7,10 d分别进行蛋白印迹标本采集[6]。灌注取脑,取双侧海马,用提取细胞膜蛋白试剂盒,用BCA法进行蛋白定量。取50 μg蛋白样品,SDS-PAGE分离蛋白,半干法转移至PVDF膜,将膜放入50 g/L脱脂牛奶中37 ℃封闭后,加入一抗(TRPC1,1 ∶1 000;NeuN,1 ∶500;β-actin,1 ∶1 000),4 ℃孵育过夜。TBST缓冲液洗膜,加辣根过氧化物酶标记的二抗(1 ∶5 000)室温孵育1 h,用增强化学放光法观察显影,凝胶成像系统拍照,用Image J软件测定条带吸光度作定量分析。
1.6 TUNEL检测皮层神经元凋亡
脑缺血模型组、SKF96365干预模型组、DMSO干预对照模型组各3只大鼠在预定的时间点深度麻醉后,经左心室灌注固定后迅速开颅取脑,40 g/L多聚甲醛溶液中固定24 h,石蜡包埋,连续冠状切片,每个脑块10张,片厚5 μm。按照TUNEL凋亡试剂盒提供步骤染色。图像分析系统采集图像,每张切片随机选取5个视野(100-200倍),计数TUNEL阳性神经元[7]。
1.7 Morris水迷宫实验(Morris water maze,MWM)
连续4 d对大鼠进行4次学习获得试验。试验持续时间最长为120 s,在此时间范围内未能找到平台的大鼠被实验者轻轻引导至平台。大鼠在连续4 d学习后,建立全脑缺血模型。干预模型组大鼠在诱导脑缺血前30 min接受侧脑室显微注射SKF96365。第1-10天在探索试验中测试脑缺血模型组和SKF96365干预模型组中的大鼠。记录大鼠在训练象限中搜索平台的时间比例,即平台的先前位置,并用作空间记忆保持的量度,以确定每组中搜索目标象限的差异[8]。
1.8 体视学分析
每个脑块以100 μm厚度垂直于长轴连续切割,并且沿着海马整个范围收集冠状切片。随机选择5个系列切片用于染色,使用系统均匀随机采样[9]。单个实验者在不知道每只大鼠条件的情况下收集该研究的所有体视学数据。使用基于计算机的Stereologer TM(SPA,Alexandria,VA)系统辅助的光学技术评估海马CA1区域中锥体神经元的总数[10]。用以下等式[11]计算锥体神经元的数量:N=(1/ssf)(1/asf)(1/tsf)ΣQ-,其中N是锥体神经元总数的估计值,并且ΣQ-是整个参考空间中计数的单元格数。ssf是截面采样分数,asf是面积采样分数,tsf是截面厚度分数。为了评估神经元数量和密度的可能差异,按照公式计算海马结构亚区神经元的总数目。
1.9 统计学分析
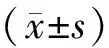
2 结果
2.1 海马神经元[Ca2+]i变化
脑缺血模型组中海马神经元[Ca2+]i在缺血后3 d时明显升高,之后逐渐降低,至10 d时降至1 d水平。SKF96365干预模型组中海马神经元[Ca2+]i虽在1 d开始上升,高于正常对照组,但其总体升高趋势被明显抑制,与脑缺血模型组相比较差异有统计学意义(P<0.05,见图1)。
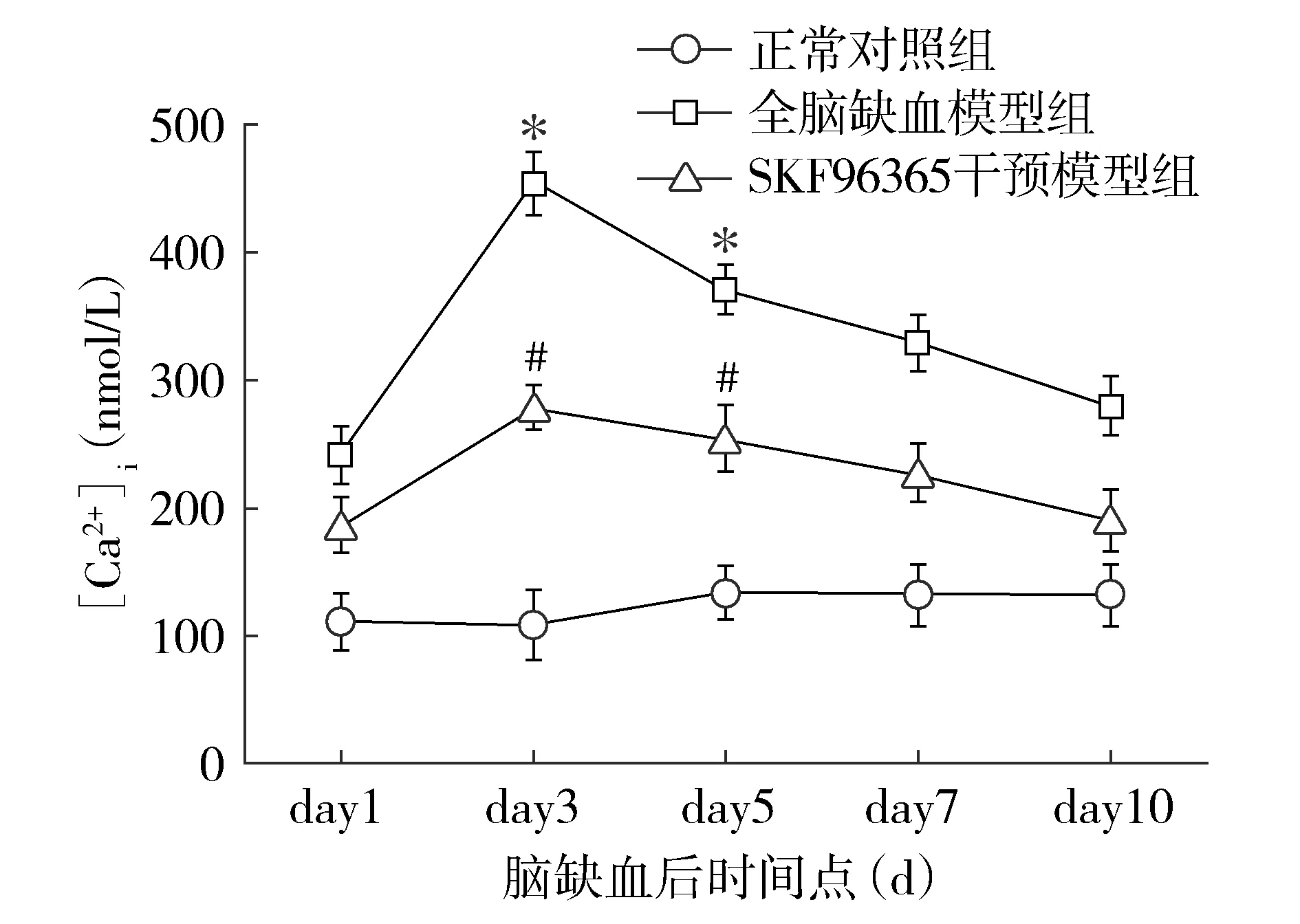
与正常对照组相比,*P<0.05;与全脑缺血模型组相比,#P<0.05图1 不同时间段海马神经元[Ca2+]i变化趋势Figure 1 Changes of [Ca2+]i in hippocampal neurons at different time points
2.2 海马神经元形态学变化
神经元特异性NeuN荧光染色可见正常对照组大鼠海马结构完整,CA1区神经元细胞荧光强度正常,脑缺血模型组海马结构基本保持完整,但CA1区神经元细胞荧光显示神经元染色有丢失(见图2A、B)。透射电镜下观察海马超微结构发现,脑缺血模型组中神经元肿胀,神经元胞内线粒体、高尔基体、内质网扩张,嵴紊乱,周围可见高密度电子物质生成,细胞核变形,部分胞质内空泡形成,染色质不均匀、边集、浓缩,可见核内凋亡小体形成;正常组中髓鞘结构完整,髓鞘形态连续,其内可见神经纤维断层;而脑缺血模型组中可见髓鞘正常结构受到破坏,髓鞘肿胀,局部有空泡形成,髓鞘连续性片层结构疏松、分离或消失,片层结构内缺乏神经纤维及线粒体。使用SKF96365阻断钙内流后可保护神经元髓鞘结构完整,维持神经纤维片层结构(见图2C-F)。
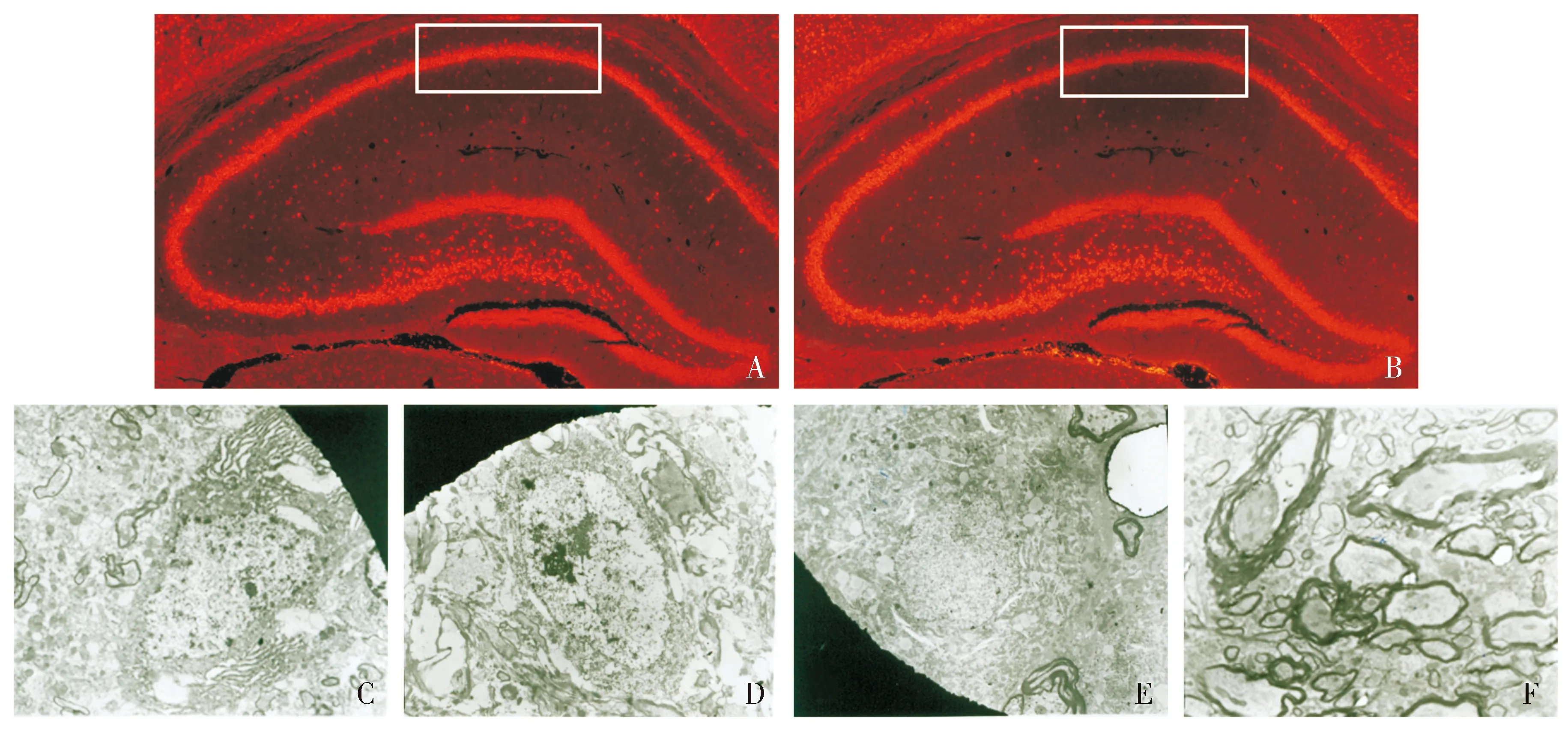
A.正常对照组大鼠海马CA1区神经元细胞荧光;B.全脑缺血模型组大鼠海马CA1区神经元细胞荧光;C.全脑缺血模型组大鼠海马神经元结构变化;D.全脑缺血模型组大鼠海马神经元核内凋亡小体,髓鞘结构破坏;E.SKF96365干预模型组大鼠海马神经元结构基本完整;F.SKF96365干预模型组大鼠海马髓鞘结构保存图2 大鼠海马NeuN荧光染色(×50)及神经元超微结构观察(×10 000)Figure 2 NeuN fluorescent staining(×50) and neuronal ultrastructure observation in rat hippocampus(×10 000)
2.3 TRPC1在海马中表达变化
Western blot结果显示,TRPC1在脑缺血后3 d表达到达高峰,之后逐渐降低,10 d时表达与1 d时相似。SKF96365干预模型组中TRPC1在3 d时表达可被抑制,其表达趋势未出现显著增加(见图3)。灰度值分析显示脑缺血模型组3 d与其他各组相比差异有统计学意义(P<0.05);NeuN在脑缺血后表达明显降低,在3 d及5 d时最显著,与其他时间段相比差异有统计学意义,之后逐渐升高(P<0.05)。SKF96365干预模型组中TRPC1蛋白的表达明显被抑制,灰度分析结果显示,各个时间段之间相比无统计学意义;NeuN在干预模型组中表达呈现逐渐增高趋势,在5 d时达到峰值,之后逐渐降低。SKF96365干预模型组中TRPC1蛋白的表达明显被抑制,灰度分析显示缺血后3 d与脑缺血模型组比,差异有统计学意义(P<0.05)。

与全脑缺血模型组相比,*P<0.05图3 脑缺血模型组及SKF96365干预模型组TRPC1在大鼠海马中的表达变化Figure 3 Expression of TRPC1 in rat hippocampus in cerebral ischemia model group and SKF96365 intervention model group at different time points
2.4 TUNEL阳性神经元表达及计数
TUNEL染色结果显示,全脑缺血模型组(见图4F-J)在1 d时已有海马神经元的TUNEL阳性细胞,之后逐渐增多,至5 d时最多;SKF96365干预模型组(图4A-E)中TUNEL阳性细胞虽较正常对照组(图4K)有增高,但其显著增高趋势被抑制。TUNEL阳性细胞计数表明,脑缺血模型组中缺血后3,5,7 d与SKF96365干预模型组对比差异有统计学意义(P<0.05,见图5)。
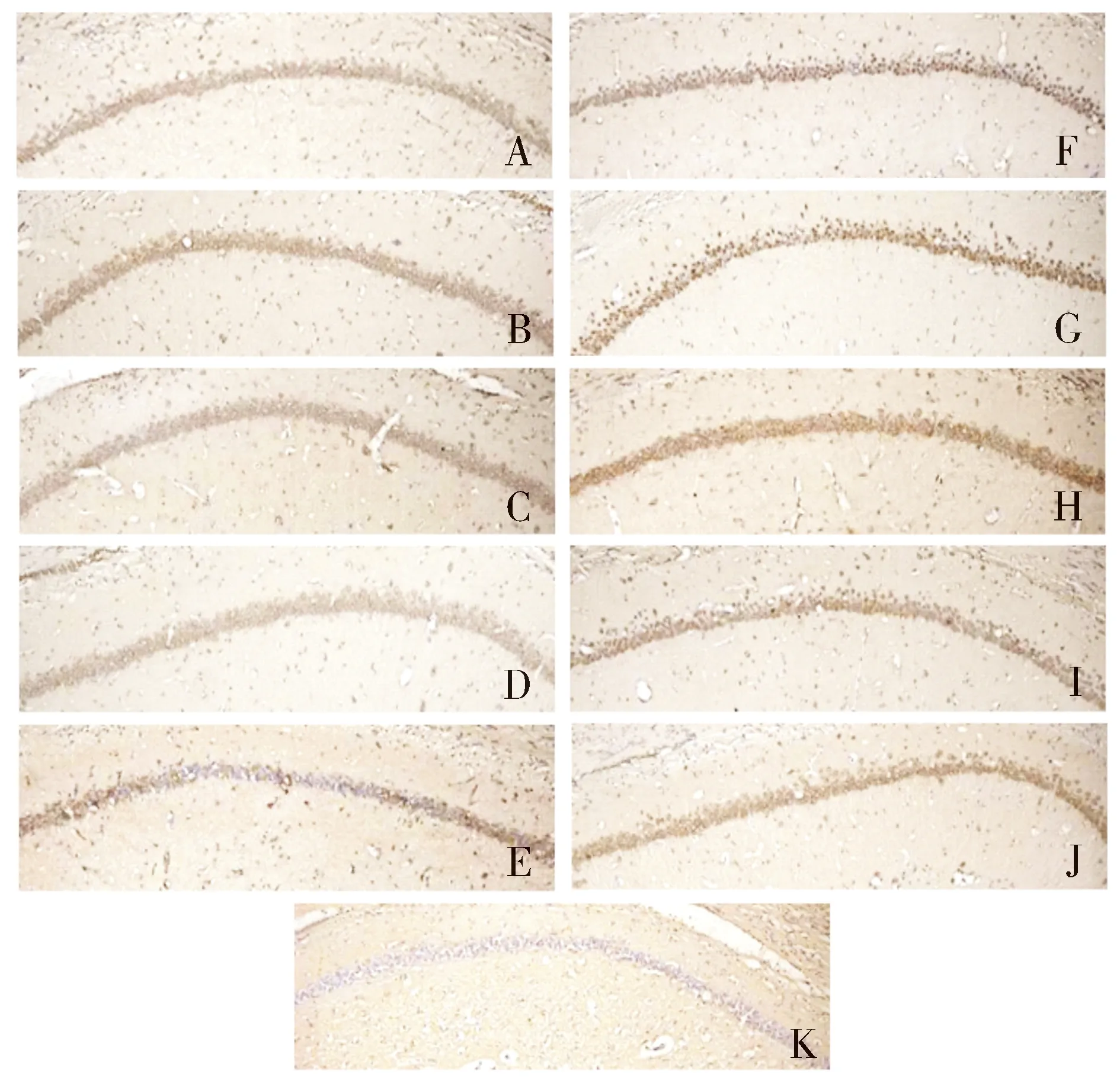
A-E.分别为SKF96365干预1,3,5,7,10 d组;F-J.分别为全脑缺血后1,3,5,7,10 d组;K.正常对照组图4 全脑缺血模型组及SKF96365干预模型组海马CA1区TUNEL阳性细胞表达变化Figure 4 Expression of TUNEL positive cells in hippocampal CA1 area of cerebral ischemia model group and SKF96365 intervention model group
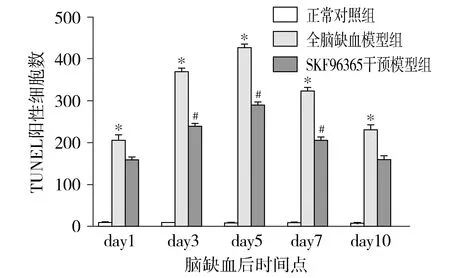
与正常对照组相比,*P<0.05;与脑缺血模型相比,#P<0.05图5 三组海马CA1区TUNEL阳性细胞数比较Figure 5 Comparison of TUNEL positive cells in hippocampal CA1 area among three groups
2.5 Morris水迷宫检测海马功能
空间搜索实验结果表明,正常对照组大鼠运动轨迹以目标象限居多;全脑缺血模型组大鼠运动轨迹,目标象限较少;SKF96365干预模型组大鼠运动轨迹目标象限较脑缺血模型组增多(见图6)。全脑缺血模型组与正常对照组相比,大鼠空间搜索目标象限的轨迹时间明显减少,差异有统计学意义(P<0.05);而SKF96365干预模型组与全脑缺血模型组相比,大鼠空间搜索目标象限的轨迹虽未能恢复至正常,但空间目标搜索时间明显减少,差异有统计学意义(P<0.05,见图6)。
2.6 体视学分析结果
大鼠海马CA1区不同视野下阳性NeuN神经元计数数量在脑缺血模型组中,5 d时为最少,之后逐渐升高;而SKF96365干预模型组的阳性NeuN神经元计数数量在5 d时与脑缺血模型组相比,数量明显增多,差异有统计学意义(P<0.05,见图7),说明存活神经元数量较全脑缺血组改善。
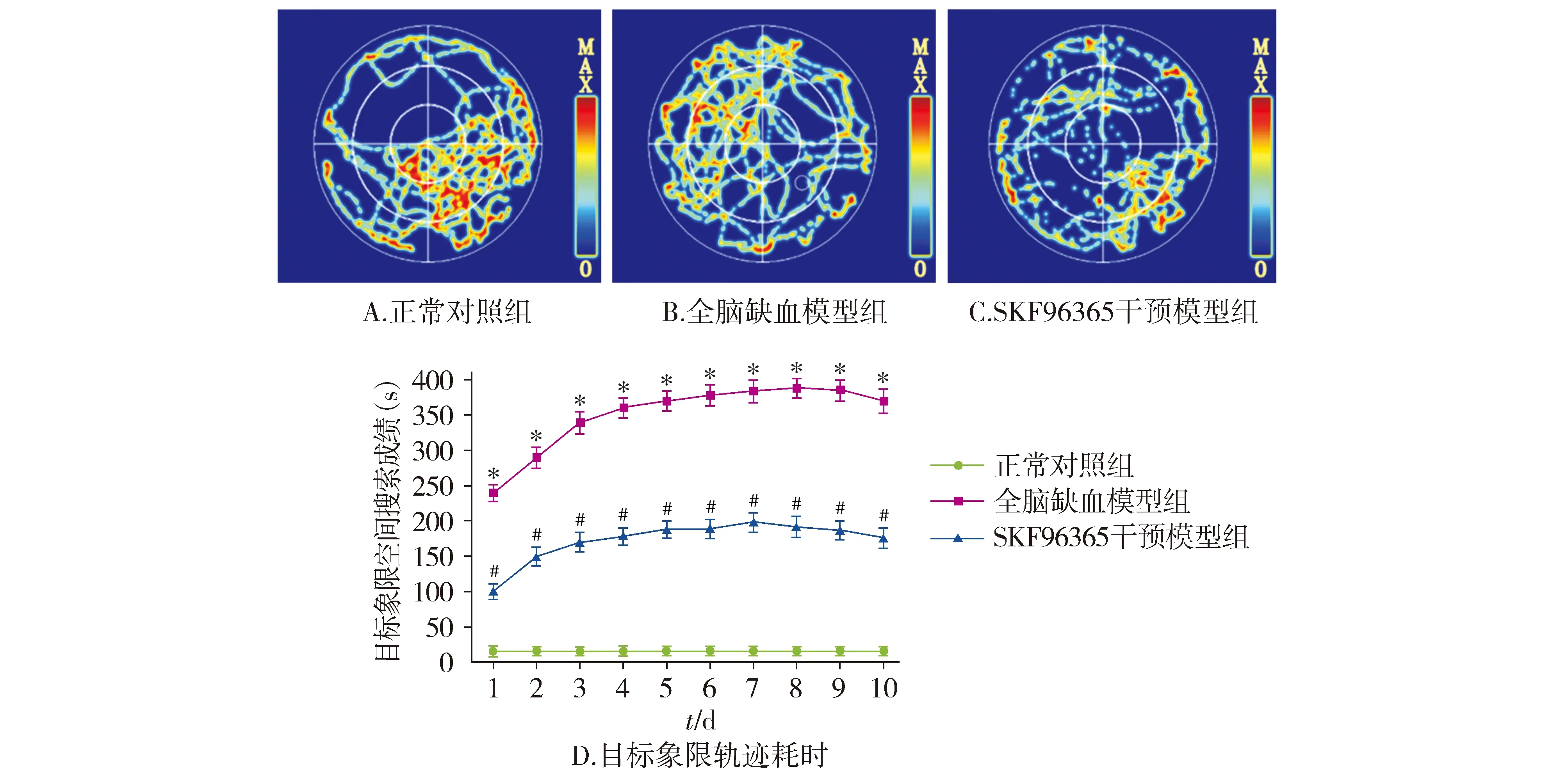
与正常对照相比,*P<0.05;与全脑缺血模型组相比,#P<0.05图6 正常对照组、全脑缺血模型组和SKF96365干预模型组大鼠在水迷宫中的运动轨迹及目标象限耗时Figure 6 The trace and target quadrant time consuming of rats in normal group, cerebral ischemia model group and SKF96365 intervention model group in water maze
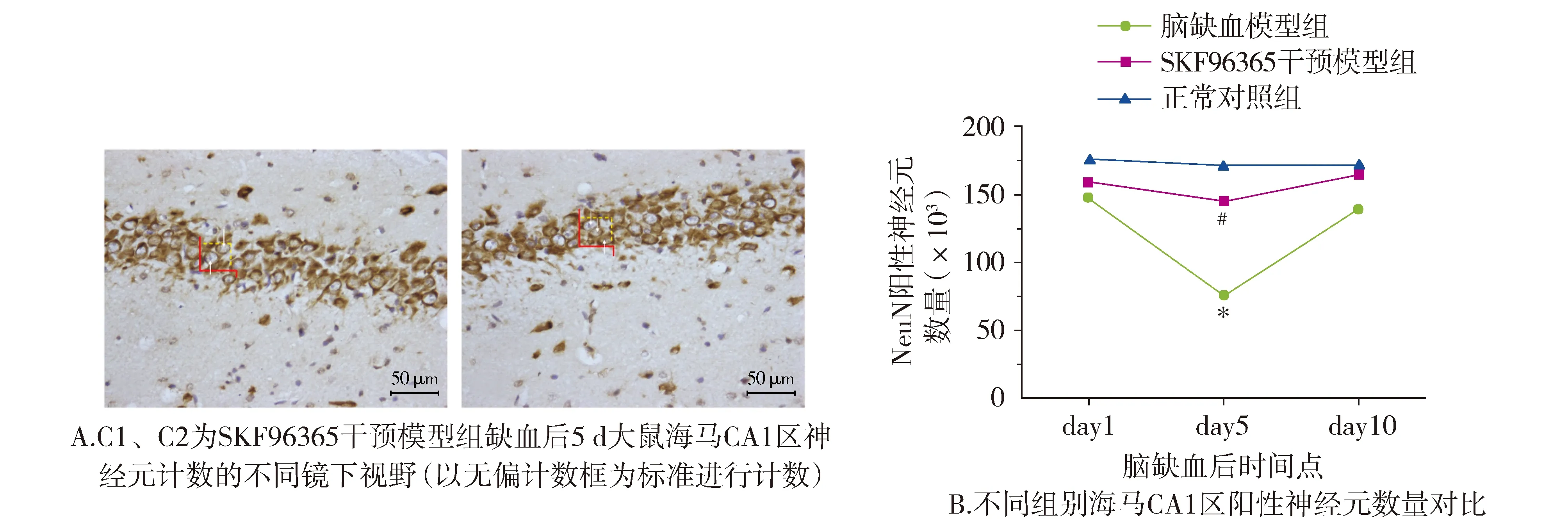
与正常对照组相比,*P<0.05;与全脑缺血模型组相比,#P<0.05图7 体视学分析海马CA1区神经元及阳性NeuN神经元数量Figure 7 Stereology analysis of the number of neurons in hippocampal CA1 area and the number of positive-NeuN neurons
3 讨论
本研究形态学研究观察到,脑缺血后的海马可见肿胀的神经元、线粒体及内质网,神经髓鞘正常结构减少或消失,可见部分核内凋亡小体,TUNEL细胞凋亡检测可见缺血后海马区神经元凋亡增加,MWM实验观察到大鼠长时程的记忆障碍,这与Pulsinelli经典四血管阻塞法建立全脑缺血模型后的海马神经元的表现相一致,说明本研究改良模型可较好模拟人脑缺血后的病理生理状态,利用此方法建立大鼠短暂性全脑缺血模型是可行且可靠的。
Ca2+是神经元最普遍的信号转导分子,而钙超载是脑缺血后导致细胞损伤最重要的一类离子失衡。近年有研究认为,位于细胞膜上的六次跨膜蛋白TRPC离子通道开放可通透Ca2+[12]。而TRPC1是其中一类阳离子非选择性钙可通透通道,在内质网膜和细胞膜上均有表达,可单独参与调控钙内流[12],亦可参与增殖和存活、分化、分泌和细胞迁移等功能[13]。TRPC1同聚体在内质网表面可形成有功能的离子通道,受胞内IP3水平的调控,参与钙池Ca2+释放[14]。
本研究观察到,TRPC1可在大鼠海马组织中广泛表达,主要表达在海马CA1区、CA2区及CA3区,脑缺血后早期海马神经元中TRPC1的表达有一过性升高,在3 d时达到峰值,之后逐渐下降,而神经元中[Ca2+]i亦在3 d时到达高峰,神经功能评分及MWM实验结果发现,脑缺血模型建立后,大鼠海马功能受损随时间逐渐加重,到3-5 d时学习记忆障碍明显加重,说明TRPC1不仅参与了脑缺血后神经元中的钙超载,而且可能是神经突触功能损伤的原因之一。本研究通过体视学分析观察到,海马CA1区神经元的数量随着脑缺血的时间推移而逐渐减少,在第5天时海马CA1区神经元数量达到最低,神经元特异性表达的NeuN阳性细胞数亦到最低,之后缓慢恢复,进一步通过TUNEL检测到神经元细胞损伤在3 d时开始形成,凋亡神经元数量逐渐升高,在7 d时最多,提示TRPC1引起的钙超载可能触发了神经元的凋亡机制,引起细胞迟发性序贯凋亡。
SKF96365是TRPC1离子通道的阻断剂之一[15],可抑制TRPC1离子通道的活性,减少Ca2+内流,在体外研究中被广泛应用[16],本研究中通过立体定向微注射的方式将SKF96365注入侧脑室,大鼠海马CA1区TUNEL阳性神经元的数量在第3,5,7天显著降低,这表明在缺血后抑制Ca2+内流会有更多神经元可得到存活。MWM实验表明,Ca2+内流减少可部分改善大鼠空间搜索的能力。进一步通过体视学分析发现,SKF96365干预模型组神经元数量在第5天时明显较脑缺血组升高,这说明,海马神经元可通过减少Ca2+内流而得到存活,突触功能可通过抑制TRPC1介导的Ca2+内流而得到改善。这与文献报道的结果相类似[17]。因此,缺血后TRPC1介导Ca2+内流可能是导致大鼠海马CA1区突触功能受损的重要原因,而脑缺血后表达升高的TRPC1可能在大鼠迟发性神经元死亡中扮演关键角色。
本研究功能学实验结果表明,在缺血后阻断过度Ca2+内流可改善海马学习记忆功能,这与TRPC1依赖性的钙库操控性钙内流在亨廷顿病小鼠模型中可影响突触稳定性及运动表现[18]相一致。文献报道,TRPC1负性调控和通道的激活还可能通过阻断小胶质细胞炎症通路的启动而发挥免疫抑制活性[19],而TRPC1的缺乏还可能通过增强Nox4衍生的活性氧生成来加剧脑缺血/再灌注诱导的神经损伤[20],同时亦可通过NF-κB介导的TRPC1表达调控自噬来诱导细胞死亡[21]。但有文献指出TRPC1蛋白缺失可导致小鼠纹状体神经元细胞凋亡以及相关蛋白质组学改变[3],因此,TRPC1在神经元细胞中的作用仍需深入研究。
综上所述,本研究表明,脑缺血后大鼠海马高表达的TRPC1在钙超载中扮演重要角色,在此过程中,过度Ca2+内流可被高表达的TRPC1所诱导,从而触发神经元死亡的许多下游机制,TRPC1可作为脑缺血后的非兴奋性中毒机制而在突触损伤中起关键作用。
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05
昆明医科大学学报(2020年11期)2020-12-28
创新作文(小学版)(2019年10期)2019-09-25
现代装饰(2018年5期)2018-05-26
中成药(2018年4期)2018-04-26
中国康复理论与实践(2015年10期)2015-12-24
中国体外循环杂志(2015年3期)2015-12-08
中国生化药物杂志(2015年4期)2015-07-07
弹箭与制导学报(2015年1期)2015-03-11
郑州大学学报(医学版)(2015年2期)2015-02-27
