
基于多组态方法研究类铍镍离子的能级与Kα跃迁参数
2019-08-01 11:03李文义豆乐乐桑萃萃
安徽师范大学学报(自然科学版) 2019年3期
李文义, 李 丹, 豆乐乐, 桑萃萃, 孙 言, 胡 峰
(1.徐州工程学院 数学与物理科学学院,江苏 徐州 221111;2.伊犁师范学院 物理科学与技术学院,新疆 伊宁 835000;3.兰州理工大学 理学院,甘肃 兰州 730050)
引 言
天体观测的快速发展需要更加准确和广泛的数据。能级、跃迁几率、线强度和振子强度等数据对于分析地面天文望远镜观测到的数据有着重要的应用,可以分析天体的组成成分、密度和温度[1]。镍元素的跃迁是目前观测到的较为常见的跃迁之一,普遍存在于太阳和其他天体发射谱中。同时,镍在惯性约束聚变中使用较为广泛,因而高离化态镍跃迁的谱线具有很大研究意义。利用天体观测设备,人们已经在太阳光、银河系中心以及超新星中观测到镍跃迁的谱线,同时对这些谱线进行了相关模拟,取得了一定成果,但是由于镍谱线强度要弱于混合的其他元素,使得镍谱线的实验分析和辨识更加复杂[2]。
本文研究的类铍的Kα跃迁参数是等离子体模拟重要研究内容之一,也是目前实验观测拟突破的内容之一。Santos等人了报道了n=3高阶的类铍能级[3]。Grumer等人报道了2s2p3P0-2s21S0的类Be跃迁特性[4]。胡峰等人报道了涵盖类Be的Kα跃迁[5]。Safronova等人用相对论多体微扰方法计算了类铍的精细结构[6];Aggarwal等人用组态相互作用方法计算了类Be镍的能级、辐射速率和碰撞激发速率[7];王凯等人用Safronova的方法计算了Z=10-30类Be的能级、波长等跃迁参数[8];Singh等人利用多组态Dirac-Fock(MCDF)理论方法计算了n=5的高阶类铍能级[9]。但上述提到的工作存在局限,例如,胡峰等人只给出了2条Kα跃迁相关信息;Singh等人并没有给出相应的Kα跃迁信息。
本文使用基于多组态Dirac-Hartree-Fock方法和相对论组态相互作用的GRASP2K原子结构程序计算拓展了以前的工作,详细讨论了1个基态1s22s2及5个激发态1s22s2p、1s22p2、1s2s22p、1s2s2p2、1s2p3的能级和相应的82条Kα跃迁。
1 理论计算方法
1.1 波函数和能级的计算
本文所用的多组态Dirac-Hartree-Fock理论方法在文献[10-11]有比较详细的介绍,这里仅作扼要的介绍。
在多组态Dirac-Hartree-Fock理论中,一个核电荷数为Z、具有N个电子的原子或离子体系的Dirac-Coulomb哈密顿量为(原子单位)
(1)

(2)
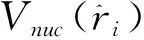
在中心场近似下单电子的旋轨波函数可表示为
(3)
式中κ为Dirac量子数,Pnk(r)和Qnk(r)分别为相对论径向波函数的大小分量,χkm为自旋函数。
N电子体系的组态波函数|Γr(PJM)〉是所有单电子旋—轨波函数组成的N阶Slater行列式波函数|Ψp〉的线性组合,即
(4)
在MCDF方法中,任一原子态α的波函数|α(PJM)〉由具有相同P,J和M量子数的组态波函数,|Γr(PJM)〉线性组合而成,即
(5)
式中nc是组态波函数的个数,Cr(α)为组态混合系数。
对角化由原子波函数(5)式构造的哈密顿矩阵,则可得到相关原子态的能量和组态混合系数。
1.2 高阶效应
Breit相互作用是由于一个横向光子发生交换而引起两个电子间库仑相互作用的修正,对于高离化态原子,Breit相互作用对原子的精细结构的影响能达到10-6。在本文的计算中,Breit相互作用的贡献是作为一阶微扰来考虑的。
MCDHF中的哈密度量不满足相对论协变条件,虽然目前对多电子离子的完整的相对论哈密顿量的研究还不清楚,但是我们采用目前国际上通用的做法,就是引入Breit相互作用来对HDC进行修正[12]:
(6)
其中,ω是两电子间交换虚光子的角频率。延迟作用和磁相互作用包含在Bω(1,2)中,所谓延迟作用就是作用传播光子的速度有限而引起的延迟效应。需要说明的是,在我们的理论计算中,按照文献[12]给出的建议,我们只考虑与角频率无关的Berit相互作用,也就是ω趋于0情况,这是因为在MCDHF计算中,可以将Bω(1,2)做微扰处理[12]。
量子动力学(QED)效应是由于电子—正电子的运动,在相互作用的过程中电磁场产生的辐射修正,包括自能和真空极化。自能是主要部分,真空极化在本计算中只考虑Uehling势。
将Breit修正和包括自能和真空极化效应的QED修正作为微扰考虑在内,可得到能量和波函数的高阶近似值。
2 结果与讨论
2.1 能 级
和文献考虑一样,通过活动空间法,将高旁观轨道纳入计算,导致组态波函数急剧增加,这对于计算的收敛性和程序本身的计算能力、计算时间要求相对提高很多,因此在保证计算精度和经济性的条件下,有必要对扩展的主量子数进行限制。本文计算选取的活动空间考虑了{1s,2s,2p,3s,3p,3d,4s,4p,4d,4f,5s,5p,5d,5f}等轨道,而5g轨道在计算中会造成计算不收敛,因此在计算中忽略了5g轨道的作用。尽管对主量子数和角量子数进行了限制,计算的结果仍然可信的。表一给出了基于MCDF方法的类Be的离子1s22s2p、1s22p2、1s2s22p、1s2s2p2、1s2p3的能级,结果包含了量子电动力学效应和Breit修正,其中量子电动力学效应考虑了两种修正:自能和真空极化。
从表1看出,真空极化和自能的贡献较小,同一组态,不同谱项之间的真空极化和自能数值相差不大。Breit贡献趋势不明显,数值上基本和自能一个量级。其中1s2p3的5S2受组态混合的影响,其能级值明显低于1s2p3的其他能级值。
为了验证当前计算结果的是可信的,在表2中比较了不同方法计算出的Be离子的1s22s2p和1s22p2能级值,其中实验值[13]来自于美国国家标准技术与研究院(National Institute of Standards and Technology,NIST),文献[9]计算结果是Singh等人利用多组态Dirac-Fock理论方法给出的,文献[14]计算结果是Landi等人用SUPERSTRUCTURE程序得出的。从表2可以看出,当前MCDHF计算结果是优于MCDF的,尤其是1s22p2的值。与SUPERSTRUCTURE程序计算结果相比,部分优于其结果。与实验值相比偏差在0.11%-0.90%,总的来说与实验值吻合较好。

表1 类Be的镍离子能级(eV)(a(b)=a×10b)

表2 不同计算方法得出的类Be能级值(cm-1),其中偏差等于abs(理论值—实验值)/实验值×100
a 偏差=(文献[9]—实验值)/实验值×100
b 偏差=(本文—实验值)/实验值×100
c 偏差=(文献[14]—实验值)/实验值×100
2.2 波 长
低密度等离子体中发射的谱线可以被认为是很好的诊断手段。因此准确的波长对于研究等离子体的状态是必不可少的。由于Kα跃迁的复杂性,使得实验观察到的波谱在小区域内相互重合,不易区分,到目前为止尚没有实验报道值,在理论方面也只有文献[5]给出了1s2s22p-1s22s2两条跃迁值。从表3可以看出,当前计算的波长值与文献[5]报道值偏差只有3×10-5nm。跃迁几率与振子强度计算值与文献[5]报道值基本一致。我们在图1和图2给出了1s22s2p-1s2s2p2和1s22p2-1s2p3的跃迁波长。为了再次验证当前计算的正确性,图1和图2纵坐标的值,来源于跃迁几率在库伦规范和哥伦布规范中表示值的比值。文献[5]指出当库伦规范和哥伦布规范中比值越是接近于1,其计算结果越是准确。从图1和图2,当前两种规范比值基本接近于1,也有部份比值偏差较大,例如1s22s2p(3P1)-1s2s2p2((1S3P)3P2)比值为1.28,这可能是由于1s2s2p2((1S3P)3P2)组态混合引起的。
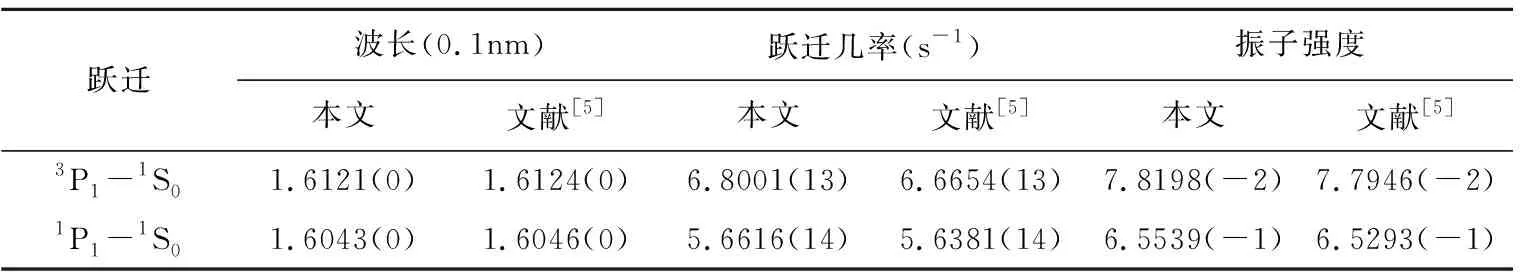
表3 1s2s22p-1s22s2Kα跃迁两种理论计算值比较
同时为了进一步验证当前计算的可信性,我们在表4给出了类Be镍离子非Kα跃迁的理论计算值和实验值。从表4可以看出,当前计算值与拟合值之间的偏差在-0.351~0.137nm之间,即当前计算值与拟合值符合很好。1s22s2p1P1-1s22p23P1和1s22s2p1P1-1s22p23P0实验报道值为32.600nm和49.820nm,需要说明的是,NIST在采用此数值时,所给的数值评估等级为最低等级E,从我们的计算结果对比来看也可以证实这一结论,因此上述两个跃迁实验值需要更加精确的测量。
3 结 语
本文在MCDHF方法的基础上,详细计算了Ni XXV Kα跃迁的能级、波长和跃迁几率,当前MCDHF的结果与已有的理论结果符合很好。这些结果对于填补类Be离子的Kα跃迁有重要的意义,同时对于指导未来开展实验也有重要的意义。
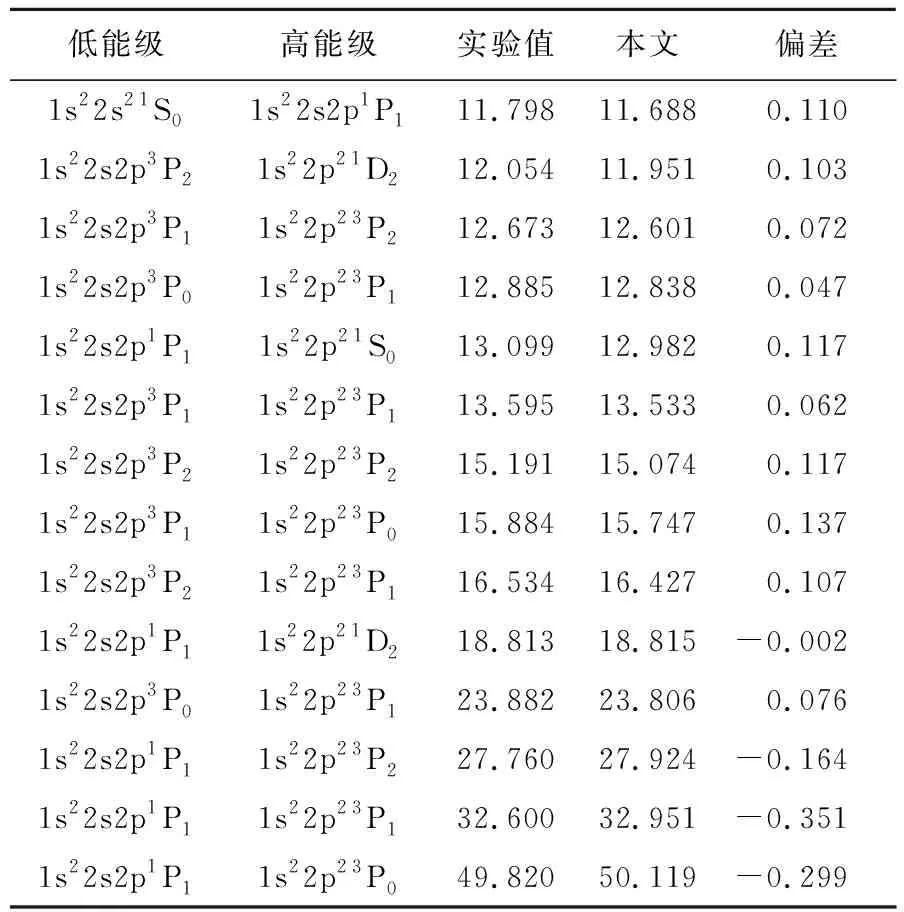
表4 类Be的非Kα跃迁波长

图1 1s22s2p-1s2s2p2Kα跃迁波长
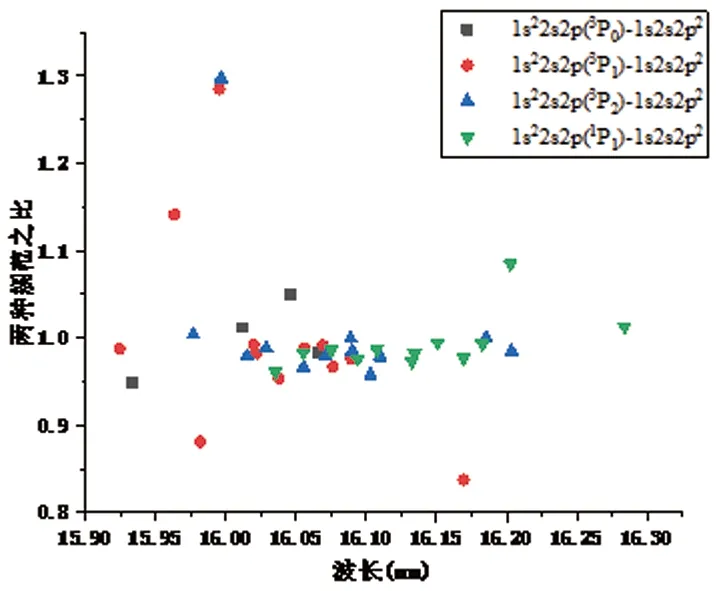
图2 1s22p2-1s2p3Kα跃迁波长
猜你喜欢
河北农机(2020年10期)2020-12-14
复旦学报(医学版)(2020年3期)2020-06-18
甘肃科技(2020年20期)2020-04-13
原子与分子物理学报(2020年5期)2020-03-17
小学生学习指导(高年级)(2017年10期)2017-09-15
凿岩机械气动工具(2017年2期)2017-07-19
小天使·三年级语数英综合(2017年6期)2017-06-07
中学生数理化·高三版(2017年1期)2017-04-20
工业设计(2016年11期)2016-04-16
中国铸造装备与技术(2015年5期)2015-12-10
