
猪链球菌2型05ZYH33菌株启动子的筛选和鉴定
2019-07-30 06:16:36黄萌萌朱金鲁刘思国张跃灵
中国预防兽医学报 2019年6期
刘 冉,黄萌萌,陈 平,朱金鲁,谢 芳,刘思国,张跃灵
(中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室/动物细菌病创新团队,黑龙江哈尔滨150069)
猪链球菌(Streptococcus suis)是一种革兰氏阳性细菌,不仅引起猪的脑膜炎、败血症和死亡,还导致人的严重感染和死亡,是一种重要的人兽共患病病原[1-3]。虽然早在1954年就发现S.suis感染,但对它的研究一直处于停滞状态。近年来,由于重复发生S.suis致死性感染,对S.suis致病因子和致病机制的研究又成为了研究热点。
目前S.suis研究所需的遗传操作工具和技术严重不足,其中有效驱动蛋白表达的内源强启动子,就是重要的遗传操作工具之一。这种启动子在高水平表达GFP用于标记菌体,以及构建无启动子基因的回复突变菌株中发挥着关键作用[4-5]。目前借鉴其它链球菌的结果,采用TufA(翻译延伸因子EF-Tu)同源蛋白的启动子PtufA,用于S.suis研究[6]。但是,一种启动子不是用于表达所有蛋白质的“灵丹妙药”,需要筛选与PtufA相当或更强的S.suis启动子,为将来S.suis致病因子和致病机制的研究提供更多的选择。
由于蛋白质的表达丰度能够反映相应启动子的强度,因此本研究通过对S.suis蛋白组分进行SDS-PAGE,选择高丰度蛋白质进行质谱鉴定,克隆相应启动子并鉴定其驱动GFP和SSU05_1921蛋白表达的能力。
1 材料与方法
1.1 主要实验材料S.suis2型05ZYH33菌株为2005年中国四川流行分离株[7];E.coli菌株MC1061F-购自上海唯地生物技术有限公司;质粒pSET2由Daisuke Takamatsu教授惠赠;Todd-Hewitt broth(THB)培养基和 Luria-Bertani(LB)培养基购自BD公司(美国);变溶菌素(Mutanolysin fromStreptomyces globisporusATCC 21553)购自 Sigma公司;PrimeSTAR Max DNA Polymerase购自宝生物工程(大连)有限公司;Cycle-Pure Kit、Plasmid Mini Kit和 Gel Extraction DNA Kit购自 Omega公司;TIA Namp Bacteria DNA Kit购自TIANGEN公司;限制性内切酶和T4 DNA连接酶购自Thermo Scientific公司;小鼠抗GFP抗体购自Abcam公司;山羊抗小鼠IgG-HRP、山羊抗兔IgG-HRP购自北京中杉金桥生物技术有限公司;兔抗SSU05_1921血清由本实验室制备并保存。
1.2 引物的设计与合成 本研究所用引物由哈尔滨博仕生物公司合成(表1)。
1.3S.suis菌体组分制备和蛋白质鉴定 取40 mL对数后期S.suis05ZYH33(OD600nm=1.0)菌体离心,收集上清液并浓缩至400 μL,该部分为分泌蛋白组分。菌体沉淀重悬于400 μL含有溶菌酶(1 mg/mL)和变溶菌素(250 U/mL)的TE-蔗糖缓冲液(50 mmol/L Tris-HCl,pH8.0,1 mmol/L EDTA,20%蔗糖)中,37℃孵育2 h,10 000 r/min离心5 min,上清部分为胞壁蛋白。沉淀重悬于400 μL PBS缓冲液,超声处理以完全裂解原生质体,这部分这为细胞膜/细胞质蛋白。上述组分进行SDS-PAGE后切取5个高表达蛋白质条带,送样BGI tech公司(中国北京),通过MALDI-TOF/TOF-MS进行蛋白质的定性鉴定。
1.4 不同启动子融合gfp或甘露糖苷酶ssu05_1921基因的载体构建 采用SoftBerry软件的BPROM程序(http://www.softberry.com/)分析获得的高丰度蛋白基因的启动子序列。采用TIANamp Bacteria DNA Kit提取S.suis05ZYH33的基因组DNA;由BGI tech公司合成gfp基因(KF410617)。利用表1中对应的引物,通过重叠延伸PCR将启动子与gfp或ssu05_1921基因融合。首先以Pro-F0177和Pro-R0177为引物,以05ZYH33基因组DNA为模板,经PCR扩增获得P0177启动子片段。再以GFP-F0177和GFP-R为引物,以合成的gfp基因为模板,扩增获得gfp0177片段。回收启动子片段P0177和gfp0177片段,混合后作为模板,以Pro-F0177和GFP-R为引物,PCR获得启动子和gfp基因的融合片段P0177-gfp。采用相同方法,利用对应引物分别经PCR扩增获得启动子和gfp基因的融合片段 P0530-gfp、P1503-gfp、P1815-gfp、P1868-gfp和P1921-gfp。融合片段经BamHⅠ和EcoRⅠ双酶切后,采用T4 DNA连接酶克隆于经同样双酶切的pSET2质粒中,分别构建pSET2-P0177-gfp、pSET2-P0530-gfp、 pSET2-P1503-gfp、 pSET2-P1815-gfp、pSET2-P1868-gfp和pSET2-P1921-gfp。ssu05_1921基因与启动子的融合同上述方法相似,采用相应引物获得融合片段,双酶切后克隆于到pSET2质粒中,构建pSET2-P0177-1921、pSET2-P0530-1921、pSET2-P1503-1921、pSET2-P1815-1921、pSET2-P1868-1921和 pSET2-P1921-1921。将上述6种重组质粒参照说明书采用化学转化E.coliMC1061F-,采用文献[8]报道的信息肽GE9诱导法转化S.suis05ZYH33。

表1 扩增gfp或ssu05_1921基因融合不同启动子所用引物
1.5 不同启动子驱动GFP在E.coli和S.suis中的表达 离心收集80 mL含有不同启动子融合gfp的E.coli和S.suis重组菌的对数晚期菌体(OD600nm=1.0),重悬于800 μL PBS中,超声裂解后,以小鼠抗GFP(1∶1 000)为一抗,山羊抗小鼠 IgG-HRP(1∶50 000)为二抗,采用western blot检测GFP的表达。取E.coli对数晚期菌液(OD600nm=1.0)于LB琼脂平板占板培养,在蓝光下观察GFP的表达情况;离心收集5 mLS.suis对数晚期菌体(OD600nm=1.0),在蓝光下观察GFP的表达情况。离心收集S.suis对数晚期菌体(OD600nm=1.0),PBS洗涤两次,然后重悬于含4%甲醛的 PBS至浓度 1×106cfu/mL。利用 CytomicsTMFC500(贝克曼)进行流式分析。以 10 μL/min的流速,通过对前向、侧向散光和侧向荧光的采集,记录50 000个事件采用CXP软件分析重组菌在S.suis中表达的平均荧光强度(AFI)。
1.6 不同启动子驱动SSU05_1921在S.suis中的表达 为了进一步评估这6个启动子的强度,本研究将1.4中构建的pSET2-P0177-1921、pSET2-P0530-1921、pSET2-P1503-1921、 pSET2-P1815-1921、 pSET2-P1868-1921和pSET2-P1921-1921 6种重组质粒分别转化S.suis,超声破碎获得全菌蛋白,并采用兔抗SSU05_1921(1∶1 000)为一抗,山羊抗兔 IgG-HRP(1∶5 000)为二抗,通过western blot检测SSU05_1921的表达情况。
2 结果
2.1S.suis05ZYH33高丰度蛋白质的鉴定 从S.suis05ZYH33中分离出3种菌体组分,即分泌蛋白组分,胞壁组分和胞膜/胞质蛋白组分。通过SDSPAGE检测,并根据蛋白丰度,同时考虑不同组分来源筛选了5个高丰度蛋白(图1)。将上述5个高丰度蛋白送至BGI tech公司(中国北京)进行MALDI-TOF/TOF-MS鉴定。质谱结果显示,分别是分泌蛋白SSU05_1815和SSU05_0177,胞壁蛋白SSU05_1868,胞内蛋白 SSU05_1503和 SSU05_0530(表 2)。巧合的是,ssu05_0530基因的启动子正是目前在链球菌中使用的强启动子PtufA,在本研究中除了对其进行鉴定外,也将其用作强启动子对照。

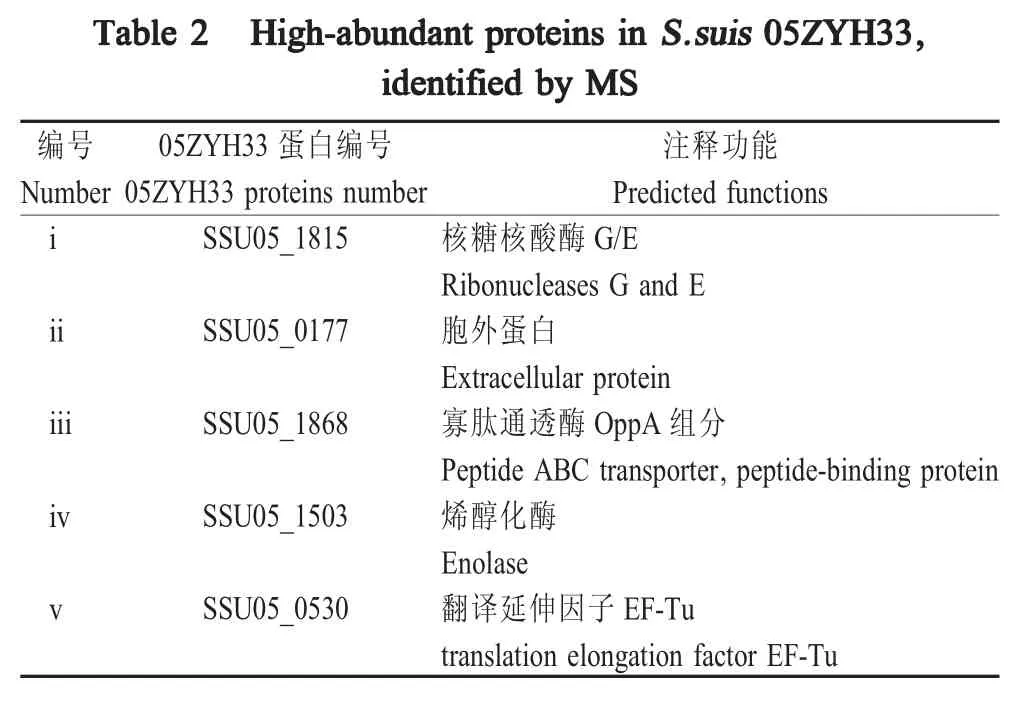
表2 质谱鉴定的S.suis05ZYH33高丰度蛋白
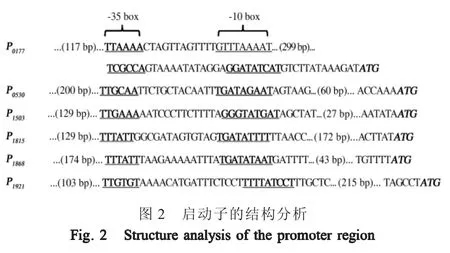
2.2 高丰度蛋白及甘露糖苷酶SSU05_1921的基因启动子分析 SoftBerry软件分析上述高丰度蛋白的基因启动子结构,结果显示,在所有基因的上游均发现了-35区和-10区,ssu05_0177基因上游则发现了两个-35区和-10区。取-35区上游至少103 bp至基因起始密码子区域为启动子序列进行克隆,对ssu05_0177基因,则将两个-35区和-10区均包括在内。根据其基因编号,这些启动子分别命名为P0177、P0530、P1503、P1815和P1868。另外,之前报道甘露糖苷酶SSU05_1921的同源蛋白在肺炎链球菌中检测不到[9],本研究在S.suis中也检测不到,初步估计ssu05_1921是弱启动子驱动(图2)。
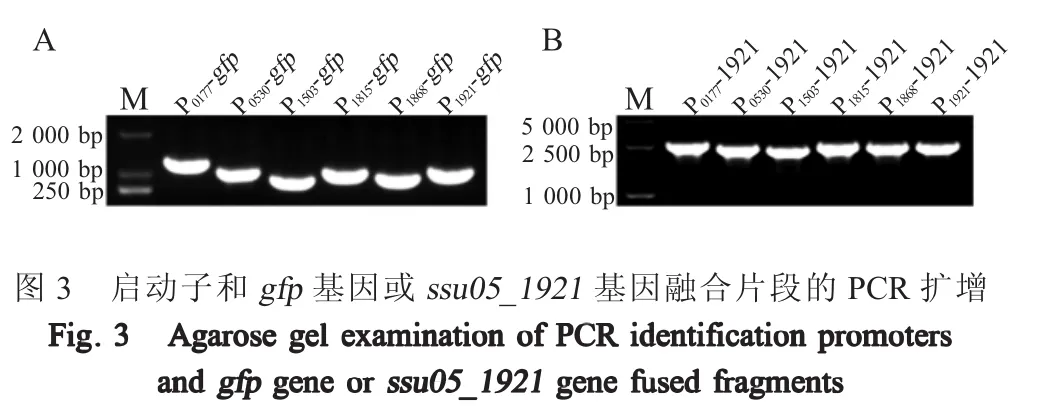
2.3 启动子与gfp基因、ssu05_1921基因的融合利用重叠延伸PCR构建融合片段P0177-gfp(1 201 bp)、P0530-gfp(1017bp)、P1503-gfp(914bp)、P1815-gfp(1069bp)、P1868-gfp(974 bp)和 P1921-gfp(1 079 bp)、P0177-1921(2 614 bp)、P0530-1921(2 430 bp)、P1503-1921(2 327 bp)、P1815-1921(2 482 bp)、P1868-1921(2 387 bp)和 P1921-1921(2 492 bp),PCR鉴定结果显示融合片段大小均与预期相符(图3)。将融合目的片段克隆于pSET2载体中,经测序分析融合位置正确,表明获得了不同启动子驱动gfp基因和不同启动子驱动ssu05_1921基因的共12种重组表达质粒。将不同启动子驱动gfp基因的6种重组质粒分别转化至E.coli和S.suis获得相应重组菌株。将不同启动子驱动ssu05_1921基因转化至S.suis获得相应重组菌株。
2.4 不同启动子驱动GFP在E.coli和S.suis中的表达 利用western blot检测不同启动子对GFP表达的驱动作用,结果显示6个启动子均可以在E.coli中驱动GFP表达,但效率存在显著差异。P1921驱动的GFP表达量非常低,难以检测,与它是弱启动子的预期符合。而高丰度蛋白的启动子P0177、P0530、P1503、P1815、P1868均能够高效驱动GFP的表达,其驱动水平均高于或相当于P0530(图4A,上)。蓝光观察与 western blot结果一致,启动子 P0177、P0530、P1503、P1815和P1868驱动的GFP表达均能够在菌体中明显表现出(图4A,下)。在S.suis中的表达情况则与E.coli中略有不同,P0177、P1503和P1868仍保持和P0530相当或更高的驱动水平,P1815驱动的GFP表达在S.suis中则显著弱于P0530,P1921驱动的GFP表达在S.suis中经western blot完全检测不到(图4B,上),蓝光观察与western blot结果一致(图4B,下)。为了进一步量化GFP表达的差异,本研究采用流式分析检测并分析了重组菌在S.suis中表达的AFI。结果显示,AFI值与每个启动子的western blot检测的强度一致,其中 pSET2-P1503-gfp/S.suis的 AFI(15.80)是 pSET2-P0530-gfp/S.suis(7.68)的两倍(图 4C)。
通过3种方法检测不同启动子驱动GFP在E.coli和S.suis中的表达,结果一致。表明P1503无论在E.coli还是在S.suis中均是6个启动子中表现最强的启动子。

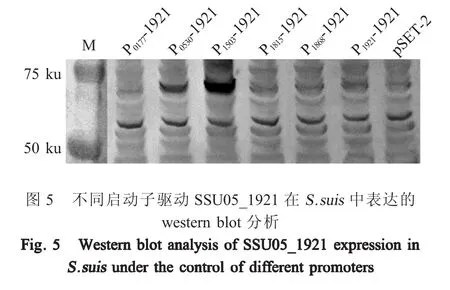
2.5 不同启动子驱动SSU05_1921在S.suis中的表达 利用western blot检测不同启动子对SSU05_1921表达的驱动作用。结果显示,即使存在基因组和质粒双拷贝的情况下,P1921也不能驱动SSU05_1921在S.suis中的表达,表明其为弱启动子。P1815驱动SSU05_1921的表达也未检测到。而P0177和P1868虽然能够驱动GFP的表达,且强度与P0530相当,但对SSU05_1921的驱动却很弱,未检测到SSU05_1921的表达。P1503和P0530均能够驱动SSU05_1921的表达,但强度存在明显差异,P1503驱动SSU05_1921的表达量高于P0530驱动其的表达量3倍以上(图5)。结果再次表明P1503是6个启动子中表现最强的启动子。
3 讨论
强启动子是很重要的遗传操作元件。为了筛选和鉴定S.suis2型05ZYH33的强启动子,本研究通过质谱鉴定到了5种高丰度蛋白。其中分泌蛋白SSU05_1815与真核转谷氨酰胺酶同源,被证明是S.suis的重要毒力因子[10]。分泌蛋白SSU05_0177 C-末端显示出与格式链球菌(Streptococcus gordonii)的毒力因子粘附素GspB有29%的同源性,提示SSU05_0177可能是S.suis的毒力因子[11]。SSU05_1868是寡肽通透酶OppA组分,介导肽的转运[12]。SSU05_0530为翻译延伸因子,在蛋白质合成中具有重要作用[13]。SSU05_1503是烯醇化酶,也称为磷酸丙酮酸水合酶,在糖酵解中发挥关键作用[14]。正是这些蛋白质的重要性决定了其在S.suis中要采用强启动子高水平的表达。
P1921被预测为弱启动子,本研究证实其在E.coli中驱动GFP表达的能力很弱,在S.suis中则完全不能驱动GFP表达,这提示它在S.suis中还有受到抑制调控的可能。这种抑制调控也体现在P1815上,因为P1815在E.coli中驱动GFP表达的能力与其它高丰度蛋白的启动子相当,在S.suis中却远远低于其它启动子。另外,同一启动子驱动不同蛋白表达的能力还存在显著不同,比如P0177、P1503和P1868在E.coli和S.suis中能够有效驱动GFP的表达,却不能驱动SSU05_1921的表达,提示需要采用不同的目的基因评估一个启动子的强弱。
对于启动子P0530,获得了看似矛盾的结果。本研究尝试了文献报道的方法,采用酶切连接P0530和ssu05_1921基因,但不能检测到SSU05_1921的表达,而采用重叠延伸PCR策略,则检测到了SSU05_1921的表达。推测可能是在P0530和ssu05_1921基因之间引入的酶切位点削弱了P0530的驱动能力,而重叠延伸策略则避免了在启动子和gfp基因之间引入任何额外的核苷酸,这提示应谨慎考虑将任何异源基因序列引入启动子和目的基因之间。
另外,本研究目前仅探究了实验室培养条件下启动子的强弱情况,虽然对体外遗传操作具有较好的指导意义,但如果用于细胞和宿主体内感染实验,还需要针对猪链球菌可能遇到的环境条件,深入研究不同条件下启动子的驱动能力。
综上,在实验室培养条件下,本研究证实P0530和P1503在S.suis中均为强启动子,而且P1503强度至少两倍于P0530。因此,除了目前使用的P0530,P1503是S.suis中强启动子的新选择。此外,由于烯醇酶在链球菌中非常重要和保守,其启动子也可能是其它链球菌中非常有潜力的遗传操作工具。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:38
煤气与热力(2021年12期)2022-01-19 05:19:30
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:24
中成药(2018年8期)2018-08-29 01:28:26
食品科学(2018年10期)2018-05-23 01:27:28
中成药(2018年2期)2018-05-09 07:20:09
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
西南医科大学学报(2015年1期)2015-08-22 13:01:46
