
体积排阻色谱在蛋白质药物聚集体领域的应用
2019-07-27 07:07:18张晓敏胡志上李红梅
分析测试学报 2019年7期
张晓敏,胡志上,李红梅*
(1.北京化工大学 生命科学与技术学院,北京 100029;2.中国计量科学研究院,北京 100013)
蛋白质药物是目前销售额最大的生物治疗药物[1],蛋白质本身非常不稳定,其结构中非共价键的稳定性很容易被外力因素( 如摇动、温度等)破坏。加热、延长保存、有机溶剂、氧气、pH值改变及其他多种因素均可引起蛋白质结构改变。作为蛋白质产品的蛋白药,在生产过程中需要进行酸处理、热处理和机械处理,这些过程中都有可能产生蛋白质折叠甚至聚集;蛋白药在贮藏、运输、销售以及用药过程中由于外力因素的作用也可能会产生聚集。蛋白质聚集现象会导致蛋白药活性降低,药品中的蛋白单体浓度降低,并可能产生有害的毒理学作用和免疫应答,甚至会发生危及生命安全的药物反应[2]。
体积排阻色谱(Size exclusion chromatography,SEC)灵敏度良好、操作简单、样品前处理少、分析速度快、分离条件温和[3],自重组蛋白药物诞生以来一直是蛋白质药物聚集体表征的金标准[4-6]。蛋白质聚集体检测技术经过多年发展,现已有20多种,常用聚集体检测技术[7-10]如图1所示。然而这些方法在检测、定量聚集体方面均有一定的局限,如:动态光散射技术只能用作初步定性工具,没有分离筛选能力;非对称流场流、分析超速离心技术定量表征聚集体不如SEC准确;纳米粒子示踪技术仅适用于粒子浓度范围为107~109颗/毫升(particles/mL)的蛋白质聚集体表征,为了达到合适的颗粒浓度通常需要对样本进行稀释,会对测量产生一定的干扰;光透法、流式成像技术在测量蛋白质样品时容易受到折射率和稀释倍数的影响,从而使测量结果不准确。综上,SEC依旧是目前表征蛋白质药物聚集体最常用的技术。该技术在监测蛋白质药物的稳定性和降解模式方面发挥着重要作用,对于指导蛋白质药物筛选及纯化工艺的开发具有重要意义。本文介绍了SEC的分离原理并对其在蛋白质药物聚集体检测中的实际应用做了归类,同时介绍了该技术在蛋白质药物聚集体应用中存在的不足及优化方法,最后对其发展前景做了简要概括。
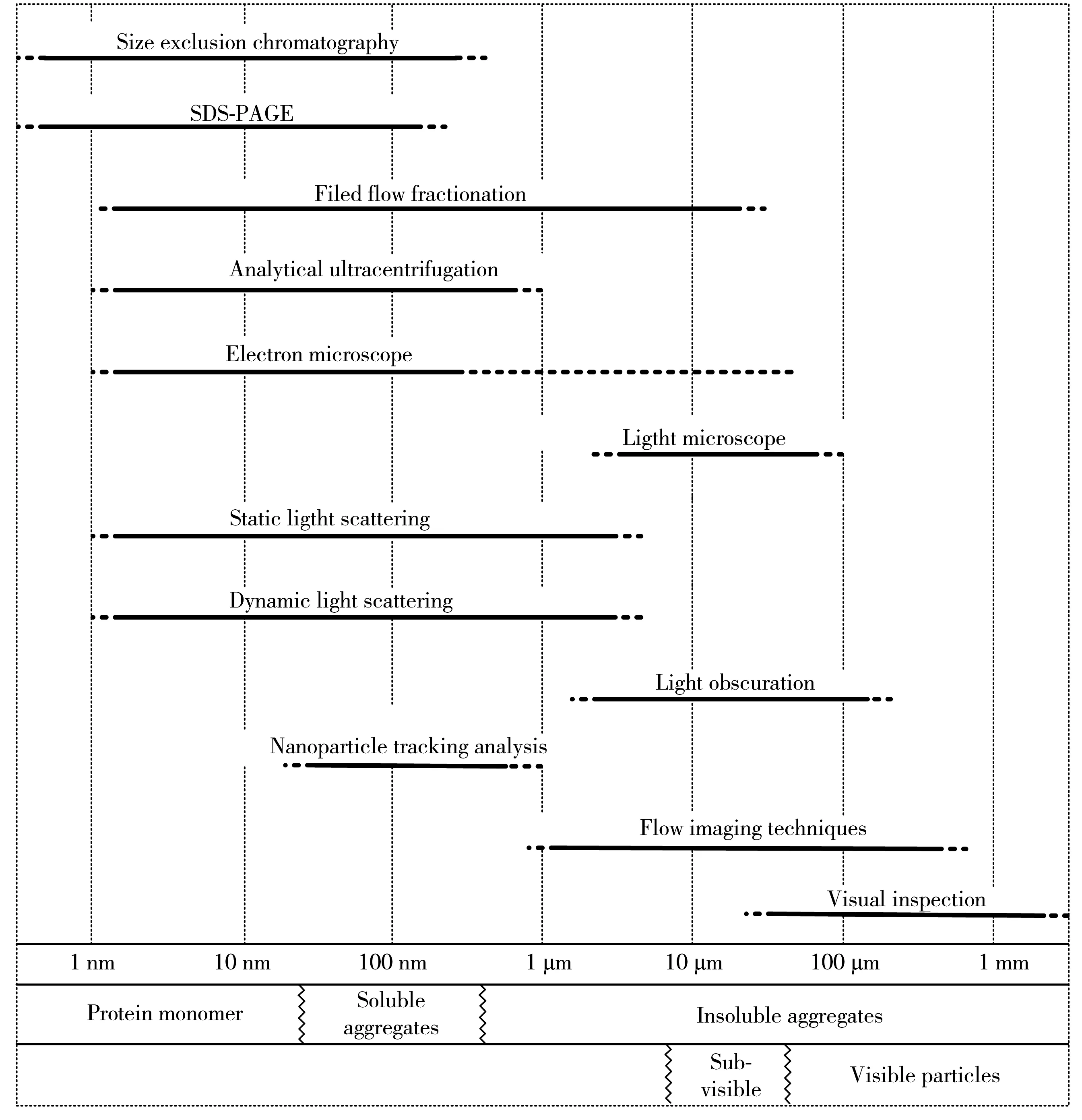
图1 检测蛋白质大小(直径)的方法及其检测范围[7]Fig.1 Methods for detecting protein size and its detection range [7]
1 体积排阻色谱原理
SEC根据流体动力学半径(Hydrodynamic radius)分离大分子化合物,其固定相由严格控制孔径的球形多孔颗粒组成,大分子化合物根据其分子体积大小差异以水性缓冲液作为流动相进行扩散[4]。Engelhardt等[6]提出以保留因子k*作为溶质在孔内滞留流动相与颗粒间流动相的比。在SEC中,该k*值是有限的,其最大值由色谱柱的孔体积(VP)和粒间孔体积(VZ)的比值给出:
(1)
(2)
式中,Velu代表从进样开始计算的通过色谱柱的实际淋洗体积。对于给定的色谱柱,其最大值k*是固定的,随着分子量增加,分子体积相应增大,溶质流经的总孔体积减少,进而导致峰展宽。目前最先进的色谱柱的最大保留因子k*约为1.6~1.8。通过引入k*,Engelhardt得出了以下半经验关系来描述色谱柱性能:
(3)
其中,H为板高;dP为粒径;DM为分子扩散系数;u为流动相线速度。
一般对于液相色谱,两组分的分离度为:
(4)
其中,tR,j、tR,i分别为两组分的保留时间;wi、wj为两组分对应的峰宽。
在SEC中由于H与u有关或与其保留体积VR有关,上述R的概念不完全适用,故引入
(5)
其中,ΔVR(ΔVR=VR,i-VR,j)为两组分保留体积之差;σ为标准偏差(与峰宽成正比);Mi、Mj为两组分的分子量(Molarmass),且Mi>Mj。当两组分的分子量差一个数量级(Mi=10Mj)时,认为两个分离度是相等的。
通常流速越低,其分离效果越好,同时分析时间越长。一般可以通过减小固定相粒径、减少色谱柱长度以及增加流速来缩短分析时间[4]。已有研究报道了柱长和流动相流速对SEC分离的影响。Popovici等[11]以1 mg/mL的四氢呋喃为流动相,聚苯乙烯混合物为样本,考察了流速为0.3、0.5、0.7、1.0、1.2、1.4、1.6 mL/min时对其分辨能力的影响,结果显示0.3、0.5 mL/min流速下的分离效果最好,当增加流速时分辨率下降,分析时间减少,且当流速大于1.0 mL/min时其分辨能力下降更快。Ricker等[12]使用ZorbaxGF-250色谱柱(250 mm×9.4 mm)研究流速对SEC分辨能力的影响时也得出相同结论,且增加色谱柱长度,分辨能力增加,因此优化SEC方法时需要综合考虑分辨率以及分析时间。
2 SEC在蛋白质药物聚集体检测中的实际应用
SEC原理简单、操作方便、灵敏度良好,常被用于质量控制[2],是评估聚集体形成的标准技术之一,在实际使用中常与多种检测器联用,如UV、RI(示差折光检测器)、多角度光散射(MALS)和粘度计等。SEC可以通过得到准确的分子量以及洞察与聚集相关的降解途径和产物来增强蛋白质表征[13],如Kiminami等[14]通过SEC联用紫外、多角度静态光散射(SEC-UV-MALS)技术评估了灭菌方法对聚合物基注射器中促红细胞生成素(EPO)储存稳定性的影响。Lancaster等[15]使用SEC联用光散射、紫外、示差折光检测(SEC-HPLC-MALS-UV-RI)的方法来表征针对艰难梭菌疫苗(Clostridium difficile vaccine)的4种不同蛋白质抗原的聚集水平。SEC联用紫外、质谱检测器(Native SEC-UV-MS)还可以表征低丰度和非共价蛋白质尺寸变体,并且为分析混合抗体药物的物理(聚合)和化学(共价修饰和碎裂)降解过程、研究抗体-抗原相互作用和其他主要类别的生物药物如Fc融合蛋白和蛋白质支架提供强大的平台[16]。SEC在监测蛋白质药物的稳定性和降解模式方面发挥着重要作用,同时也被用来研究蛋白质药物的聚集机制。
2.1 在蛋白质药物稳定性方面的应用
Al-Ghobashy等[17]将SEC作为一种技术手段研究了pH值对蛋白质药物稳定性的影响,结果表明蛋白质药物的降解与溶剂的pH值有关,在较低pH值时蛋白质药物降解程度最低。抗体药物偶联物(Antibody-drug conjugate,ADC)将单克隆抗体的特异性靶向特性与小分子药物的极高效能相结合[18-19],是复杂的蛋白质药物治疗剂。ADC由3部分组成:单克隆抗体、细胞毒性药物以及将抗体和小分子药物连在一起的接头。Li等[20]将SEC与在线二维色谱中的反相色谱(RP-HPLC)联用,用于ADC药物制剂在不同温度和配方pH值下稳定性的研究。该技术可直接在ADC样品中鉴定和定量未结合的小分子药物和相关的小分子杂质,其中SEC方法在第一维不仅能将小分子杂质与完整的ADC分开,而且还能提供关于ADC的尺寸(单体、二聚体、多聚集体等)二维信息。
2.2 在抗体包装产品稳定性检测中的应用
长久以来,药物注射器大部分采用玻璃制成。已有研究表明玻璃注射器润滑所需的硅油会引起某些蛋白质(包括蛋白质药物)的聚集[21-23],目前其与蛋白质的相互作用仍然是一个热门研究领域。Felsovaly等[24]发现来自针筒的硅油滴会干扰蛋白质聚集体的定量和表征。SEC作为检测蛋白质聚集体的常用技术,被用于预填充注射器以及其他抗体药物包装中蛋白质药物稳定性的研究。如Waxman等[25]运用SEC作为其中一种技术手段研究几种蛋白质样品在静态条件下和受到机械应力刺激时存储在由硅化玻璃以及无硅塑料制成的预填充注射器中蛋白质的稳定性。结果表明无硅塑料制成的注射器中蛋白质聚集体更少。Sugimoto等[26]在评估外部配药药房用一次性玻璃瓶和玻璃安瓿瓶中重新包装的Avastin®(贝伐单抗)的稳定性和功效研究中,使用SEC来定量重新包装的贝伐单抗样品单体和可溶性聚集物水平,结果表明贝伐单抗在安瓿瓶和一次性玻璃瓶中复配后保持稳定,没有观察到显著的聚集、片段化或生物活性丧失。
2.3 聚集机制研究
SEC作为一种技术手段也被科研人员用来研究蛋白质的聚集机制。Xiong等[27]在研究阿尔茨海默病相关的Aβ(β-淀粉样蛋白)肽Aβ(25~40)的聚集机制中将SEC作为其中一种技术手段,评估样品的初始聚集体分布。Paul 等[28]通过SEC技术发现4号单抗株(mAb4)及其Fab片段的雾化导致其单体含量显著降低,Sadavarte等[29]在研究中采用SEC和疏水相互作用膜色谱确定单克隆抗体寡聚体解聚的程度,结果显示热循环导致样品二聚体分解。
3 SEC在蛋白质药物聚集体分析中存在的不足及优化方法
SEC方法灵敏度良好、重复性强、检测速度快,在蛋白质药物聚集体分析中得到了广泛使用。但其在实际应用中仍存在一定的不足,主要表现为蛋白质药样品与SEC固定相之间的非特异性相互作用[30]会导致洗脱延迟,色谱峰拖尾,基线漂移,蛋白质回收率低等问题,其次,由于SEC检测范围有限,而蛋白质药物形成的聚集体尺寸范围较大(从简单的二聚体到可以被SEC色谱柱过滤掉的大的多聚体),导致检测结果不准确。以下将针对这两个问题做简要探讨。
SEC用于蛋白质药物分析时,蛋白质药物聚集体比单体更容易与柱基产生非特异性相互作用,导致分析不准确、可能检测不到对产品安全性或功效有显著影响的聚集体[31]。通常,新的SEC色谱柱会更容易吸附蛋白质,且初次进样会比多次进样吸附蛋白严重。其次,蛋白质药物的配方即溶剂和SEC流动相并不完全适配,当蛋白质药样品被注入与其成分不同的流动相时,会增加蛋白质样品与SEC柱的非特异性吸附,也会对其非共价聚集体的分布造成一定的干扰,导致测量不准确。因此选择合适的流动相以及低吸附的色谱柱对于SEC方法优化是必须的。
但随着SEC色谱柱填料的发展,一些研究表明,在采用新型填料时,蛋白质样品与柱基的非特异性相互作用基本可以忽略。如Goyon等[32]在研究钠、钾添加剂对SEC分离蛋白质药物与聚集体的影响时,分别采用了安捷伦公司(Agilent Technologies)的AdvanceBioSEC色谱柱、日本Tosoh(Tokyo,Japan)公司的TSKgel UP-SW3000色谱柱以及美国Waters(Milford,MA,USA)的ACQUITY UPLC BEH200色谱柱,通过SEC耦合在线荧光光谱法测定,发现10个商业单克隆抗体的聚集水平在3个不同的UHP-SEC柱上相似,表明3个SEC色谱柱填料对蛋白质药物聚集体影响低,其非特异性相互作用可以忽略不计。因此本文侧重于讨论流动相的优化。
3.1 流动相的优化
一般情况下,流动相低到中的盐浓度可降低蛋白质药物样品与SEC固定相的相互作用,使蛋白质样品洗脱时间正常且回收率提高,高浓度盐则会导致蛋白质盐析。如Kamberia等[33]使用TSK-G2000SW色谱柱以人甲状腺激素(Human parathyroid hormone,hPTH)(1~34)为样品分析了NaCl浓度对样品非共价聚集体回收率的影响。结果表明,增加盐浓度且当NaCl为100 mmol/L时,其回收率达到最大(78.2 %),NaCl为600 mmol/L时,由于盐析,样品回收率降为0。Ricker等[12]使用ZorbaxBioSeries GF-250(250 mm×9.4 mm 或 250 mm×4.6 mm)色谱柱研究了不同盐浓度流动相对3种抗体的影响,其中3种抗体最佳盐浓度为0.1~0.2 mol/L,当盐浓度<0.1 mol/L时,样品回收率快速增加,盐浓度为0.1~0.4 mol/L时达到平缓,当盐浓度大于0.4 mol/L时回收率快速降低。保留时间在该盐浓度范围内无显著变化。
氨基酸在远紫外区均有光吸收,目前很少用于SEC流动相中,但一些研究表明氨基酸可以减少或抑制蛋白质样品与硅胶柱基的非特异性相互作用,如精氨酸。Yumioka等[34]使用两根新的TSKgel G3000SWXL色谱柱,分别以pH 6.8的0.2 mol/L NaCl缓冲液以及0.2 mol/L(arginine HCl)精氨酸缓冲液为流动相,对比了鼠源抗体mIgG1在相同流速、相同进样量条件下的分离度以及回收率。结果表明,在精氨酸缓冲溶液中蛋白样本单体和二聚体的分离度优于氯化钠缓冲溶液(精氨酸缓冲溶液中样品单体和二聚体分离度为1.36,氯化钠缓冲溶液中为1.25),同时该样品在精氨酸缓冲溶液中的回收率优于氯化钠缓冲溶液中,该研究还发现样品在精氨酸缓冲溶液下的聚集体含量显著高于氯化钠缓冲溶液。
加入去垢剂也可以抑制蛋白样品与固定相的相互作用,但由于去垢剂会与蛋白样品结合,可能破坏蛋白样品的非共价聚集体,所以一般很少在SEC分析蛋白质药样品[31]中使用。微量的去垢剂是否可以在不破坏非共价聚集体的情况下抑制蛋白样品与柱基的吸附作用还有待进一步研究。
3.2 检测范围难以覆盖整个蛋白质聚集体
蛋白质药物聚集体通常被认为是产品降解产物,尺寸范围包括纳米级的二聚体以及数百微米肉眼可见的颗粒[35]。聚集是影响蛋白质药物安全性和功效的重要问题,并与蛋白质免疫原性有关[7]。因此需要准确定量表征蛋白质聚集体以满足临床要求。SEC由于自身条件,如色谱柱填料、色谱柱长等导致检测范围存在一定限制,为了确保该技术检测蛋白质药物聚集体的可靠性以及准确性,本文提出几种常用的检测手段,作为SEC验证时的补充技术。
3.2.1 与聚丙烯酰胺凝胶电泳(SDS-PAGE)或十二烷基硫酸钠毛细管电泳(CE-SDS)技术联合使用SDS-PAGE是一种非常常见、相当稳健的方法,该方法操作简单,可以提供样品近似分子量和数量信息。SDS的存在意味着非共价聚集体被破坏,所以该方法仅可检测共价聚集体。如果使用还原条件,SDS-PAGE可以区分由二硫键连在一起的聚集体和由其他(不可还原的)共价键连在一起的聚集体。目前SDS-PAGE正在被更适合量化的毛细管电泳CE-SDS所取代。使用CE-SDS可以自动运行样品并通过紫外吸收而不是染料结合进行定量,实现与SDS-PAGE类似的结果。SEC和SDS-PAGE和/或CE-SDS的组合通常是质控分析的一部分[2]。如Turner等[36]使用SEC、CE-SDS、动态光散射(DLS)以及流式成像技术(FI)等技术对NIST单克隆抗体进行大小变异体监控,其中优化过的CE-SDS以及SEC技术可涵盖大小约为100 nm及以下的大小变异体,在监视抗体结构、纯度及稳定性方面发挥了重要作用。Dada等[37]对比了SEC与CE-SDS技术在监测IgG1单克隆抗体降解中的应用,研究表明CE-SDS可作为SEC在监测抗体铰链区片段化中的替代技术,同时两种技术相结合可以获得抗体片段化较为全面的表征。Buecheler等[38]在其研究中采用SEC与非还原状态下的SDS-PAGE技术检测酿脓链球菌来源的免疫球蛋白G降解酶(Immunoglubulin G-degrading enzyme of Streptococcus pyogenes,IdeS)对IgG1单克隆抗体的消化速率,结果表明使用IdeS几乎可以使该实验中IgG1单克隆抗体完全消化(消化蛋白量> 98%)。
3.2.2 与分析超速离心法(Analytical ultracentrifugation,AUC)技术联合使用AUC是表征大分子沉降行为和溶液中聚集体存在的有力技术,主要应用沉降速度模式(SV-AUC)。AUC的主要优点之一是蛋白质样品通常可以在其样品缓冲液中无需样品前处理进行表征。减少了由样品制备、稀释或基质效应引起的样品中聚集体的形成或破坏。由于没有目标分子与色谱柱或膜的相互作用,AUC是定性交叉核对或检验由SEC 获得数据准确性的有利工具[2]。 AUC可用于验证样本中可能存在的聚合类型是否被SEC漏掉。Philo[9]发现用SEC检测的单体峰如果用SV-AUC分析,则包含单体和二聚体。Gervais等[39]将AUC与SEC技术联合使用,在对碱性蛋白质高聚物进行定量分析的过程中发现,SEC方法开发中应评估碱性蛋白质与柱基的非特异性结合作用,以确保对较大聚集物的分辨能力不受影响。
3.2.3 与动态光散射(DLS)技术联合使用DLS(Dynamic light scattering)是一种用于测定粒径在1~2 nm到3~5 μm范围内粒径分布的非破坏性技术[2]。该技术一般用于蛋白质药物稳定性研究。Al-Ghobashy等在SEC与DLS技术检测pH值对生物制药稳定性影响的研究中,使用DLS技术观察到了SEC色谱图中未观察到的高分子聚合物的存在[17]。
3.2.4 与流场流分离(Flow field-flow fractionation(F4))技术联合使用F4最近已被美国食品药物管理局(FDA)批准为蛋白质产品验证中SEC的补充技术[40],其小型化中空纤维流场分离(Hollow fiber flow field-flow fractionation,HF5)技术显示了以下优点:检测范围较宽、分离机理温和、稀释倍数低和灵敏度高。为此Marassi等[40]提出将HF5作为评估单克隆抗体样品中聚集体含量的补充技术,并对SEC和HF5性能进行了比较研究,证明了SEC和HF5之间的互补性以及它们被用作蛋白质药物表征和质量控制方法的可行性。
3.2.5 与其他多种方法联合使用SEC灵敏度高、操作简单、样品前处理少、分析速度快、分离条件温和[3],自重组蛋白药物诞生以来一直是蛋白质药物聚集体表征的金标准。然而在实际应用过程中仍存在由于蛋白质与固定相的非特异性相互作用所导致的洗脱延迟、色谱峰拖尾、基线漂移、蛋白质回收率低等问题。其次,由于该技术分离大分子化合物的尺寸范围有限,而蛋白质聚集体大小范围包括纳米级的二聚体以及数百微米级的肉眼可见颗粒,因此需要结合多种技术来获得有关蛋白质聚集体形成方式及其结构、大小等的全面信息。He等[41]开发了SEC与混合模式液相色谱(LC)在线偶联的方法,该方法允许同时测量蛋白质药物中各种组分,包括活性药物成分(蛋白质)和各种赋形剂,如阳离子、阴离子、非离子疏水表面活性剂和亲水性糖类等。Li等[20]开发了一种使用SEC和RP-HPLC二维色谱模式的技术,用于直接分析抗体药物偶联物(Antibody drug conjugates,ADC)中的游离药物和相关小分子杂质。近年来不断涌现的分析仪器及方法也为SEC与其他多种方法联合使用,从而实现蛋白质聚集体全面表征提供了可能。如Hu等[42]在实验室搭建了一款新的流式细胞仪(FC,Flow cytometry),该仪器可以轻松检测低至200 nm左右的标准聚苯乙烯微球以及微米级别的二氧化硅颗粒,通过该仪器对比不同加热时间、加热温度下单克隆抗体的聚集情况,结果表明该仪器可以检测到大小为0.2~10 μm的蛋白质聚集体,该技术为蛋白质聚集体表征技术的选择提供了新的参考。Yoneda等[43]比较研究了定量光散射技术(Quantitative laser diffraction,qLD)与共振质量测量技术(Resonant mass measurement,RMM)、纳米粒子示踪技术(Nanoparticle tracking analysis,NTA)、流式成像技术(Flow imaging,FI)以及光透法(Light obscuration,LO)在蛋白质聚集体定量表征中的应用,发现qLD和RMM 在粒径范围0.3~2 μm之间时数据具有良好一致性,同时qLD与FI技术在粒径范围2~20 μm之间时数据具有良好一致性,从而为蛋白质聚集体表征技术的组合提出了新方法。
4 结语与展望
综上所述,SEC目前仍然是蛋白质药物开发、质量控制和稳定性研究中最常用的方法。该技术适用于1~50 nm范围内聚集体的分析。当不引起样品聚集状态的变化且可消除与柱填充介质的非特异性相互作用时,SEC被认为是稳健且准确的。
猜你喜欢
合成化学(2024年3期)2024-03-23 00:56:44
合成化学(2023年12期)2024-01-02 01:02:18
肝博士(2022年3期)2022-06-30 02:48:48
河南工业大学学报(自然科学版)(2021年6期)2022-01-26 06:36:08
海外星云(2021年9期)2021-10-14 07:26:10
青苹果·教育研究版(2016年9期)2016-12-23 11:52:36
兽医导刊(2016年12期)2016-05-17 03:51:50
肝博士(2015年2期)2015-02-27 10:49:44
现代检验医学杂志(2015年4期)2015-02-06 02:02:06
石油化工(2014年1期)2014-06-07 05:57:08
