
超高效液相色谱法测定大鼠血浆中左氧氟沙星质量浓度*
2019-07-17 05:56邱妍川杨宗发
中国药业 2019年14期
邱妍川 ,刘 阳 ,钟 玲 ,杨宗发 △
(1.重庆医药高等专科学校,重庆 401331; 2.重庆药友制药有限责任公司,重庆 401121)
左氧氟沙星可用于治疗呼吸道、泌尿道、皮肤和软组织感染[1-3]。其市售口服剂型以片剂、胶囊剂为主,将其制成新剂型胃漂浮缓释微丸,可延长药物在胃中的停留时间,使药物在胃中充分释放,提高胃部疾病的局部疗效;同时,因该剂型可增加药物在胃及小肠上部的吸收,稳定血药浓度,故可提高其口服生物利用度[4-6]。为进一步提高该制剂疗效,本研究中给予大鼠灌胃自制左氧氟沙星胃漂浮缓释微丸,并建立了测定大鼠血浆中左氧氟沙星质量浓度的超高效液相色谱(UPLC)法。现报道如下。
1 动物、仪器与试药
1.1 动物
清洁级SD大鼠,雌雄各半,6周龄,体质量为150~200 g,饲料为基础饲料,由重庆医科大学实验动物中心提供,使用许可证号为SYXK(渝)2017-0023。
1.2 仪器
1290型超高效液相色谱仪(美国Agilent公司);XS205DU型十万分之一电子天平(瑞士梅特勒托利多公司);IKA Vortex Genius 3型涡漩混合仪(德国IKA公司);移液器(德国Eppendorf公司);Allegra X-22R Centrifuge型多功能台式高速低温离心机(美国Beckman公司)。
1.3 试药
左氧氟沙星对照品(批号为130455-201607,纯度97.3%),环丙沙星对照品(批号为130451-201203,纯度84.2%),均购自中国食品药品检定研究院;左氧氟沙星原料药(浙江国邦药业有限公司,批号为160809,纯度>99%);左氧氟沙星胃漂浮缓释微丸(自制);甲醇、乙腈均为色谱纯,磷酸二氢钾为分析纯,水为去离子水。
2 方法与结果
2.1 色谱条件
色谱柱:WatersACQUITYUPLC BEH C18柱(50 mm×2.1 mm,1.7 μm),配置同填料类型的保护柱;流动相:0.05mol/L 磷酸二氢钾缓冲液-乙腈(75∶25,V/V);流速:0.3mL/min;柱温:30℃;进样量:1μL;检测波长:294nm。
2.2 溶液制备
称取左氧氟沙星对照品20.55 mg,精密称定,置100 mL容量瓶中,加入适量甲醇,超声使溶解,再用甲醇定容,摇匀,作为对照品溶液。称取环丙沙星对照品10.12 mg,精密称定,置100 mL容量瓶中,加甲醇溶解并定容,摇匀,作为内标贮备液,精密移取5 mL置50 mL容量瓶中,加甲醇定容,作为内标溶液。
2.3 血样处理
取6只SD大鼠,试验前禁食12 h,灌胃自制左氧氟沙星胃漂浮缓释微丸(40 mg/kg)。分别于给药后0.25,0.5,1,2,4,8,12,24 h 时眼眶取血 0.3 mL,置肝素化塑料离心(EP)管中,5000r/min 离心 10min,取血浆,置-20℃冰箱中低温保存。精密量取大鼠血浆100 μL,置1.5mLEP管中,加入内标溶液50μL和甲醇300μL,涡旋0.5 min,1.2×104r/min 离心 5 min,取上清液,氮气吹干,加入流动相100μL使残渣溶解,即得空白血浆样品。
2.4 方法学考察
专属性试验:分别取空白血浆样品,空白血浆+对照品+内标,给药12 h后的血浆样品+内标进样,记录色谱图。结果见图 1。可见,左氧氟沙星与内标环丙沙星分离良好,保留时间分别为3.0 min和3.9 min,血浆中内源性物质不干扰左氧氟沙星的分离和测定,表明专属性良好。

图1 超高效液相色谱图
线性关系考察:精密量取不同质量浓度的左氧氟沙星对照品溶液各50 μL,置不同的1.5 mL EP管中,氮气吹干,加入空白血浆100 μL,配成含左氧氟沙星分别为 0.20,0.50,1.00,5.00,10.00,12.00,15.00 μg/mL的含药血浆样品,照2.3项下方法处理样品后按2.1项下色谱条件进样分析,以血浆质量浓度(C)为横坐标、左氧氟沙星与内标的峰面积之比(R)为纵坐标进行线性回归,得回归方程R=12.018C+0.848,r=0.999 4(n=7)。结果表明,血浆中左氧氟沙星质量浓度的线性范围为0.20~15.00 μg/mL。大鼠血浆中左氧氟沙星检测限为 0.20 μg/mL,RSD为 4.6%(n=7)。
相对回收率试验:分别用2.3项下处理好的空白血浆样品与对照品溶液配制高、中、低质量浓度和定量限质量浓度(12.00,5.00,0.50,0.20 μg/mL)的血浆样品,按2.1项下色谱条件进样分析,按线性回归方程计算各质量浓度的实测值。质量浓度实测值除以理论值的百分比即为相对回收率。结果见表 1。
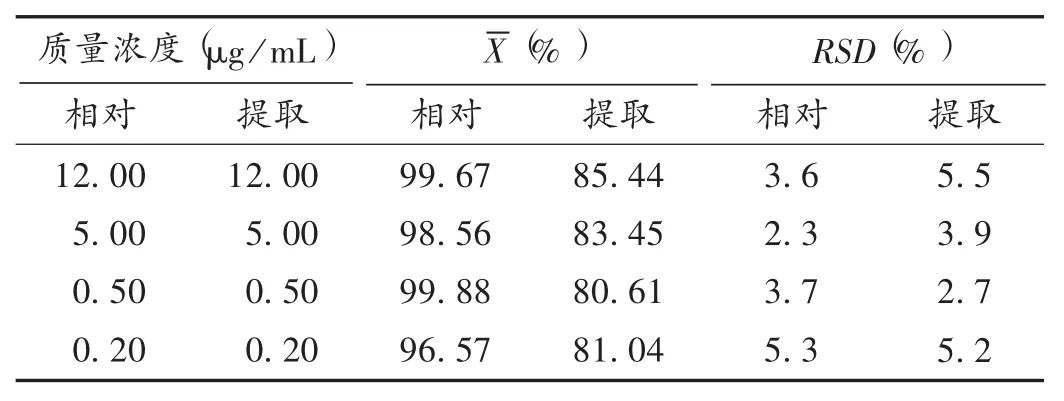
表1 回收率试验结果(n=6)
提取回收率试验:精密量取不同质量浓度的左氧氟沙星对照品溶液50 μL,置不同EP管中,氮气吹干,加入空白血浆100 μL,按2.3项下方法处理,制得高、中、低质量浓度及定量限质量浓度水平(12.00,5.00,0.50,0.20 μg/mL)的提取回收率供试品溶液。按2.1项下的色谱条件进样分析,结果平均提取回收率介于80.61%~85.44%(n=6),详见表 1。
精密度试验:精密吸取空白血浆100 μL,置1.5 mL EP管中,加入对照品贮备液及甲醇适量,制成含左氧氟沙星分别为 12.00,5.00,0.50,0.20 μg/mL 的高、中、低及定量限4个质量浓度水平的血浆样品,每个质量浓度配制6份,按2.3项下方法处理,进样1 μL,同一样品,1 d内测定6次,计算日内变异;同一样品,每天测定1次,连续测定6 d,计算日间变异。结果见表 2。

表2 精密度试验结果(n=6)
稳定性试验:依法制备高、中、低质量浓度的血浆样品,分别考察室温放置12 h,-20℃条件下冷冻12 d,血浆样品3次冻融后再处理的稳定性。结果显示,测得左氧氟沙星质量浓度与真实质量浓度的偏差均小于15%,表明样品在3种条件下均能保持稳定。详见表 3。
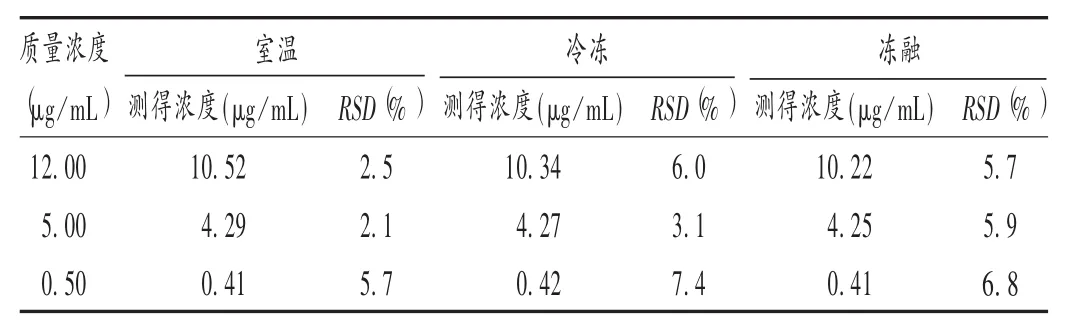
表3 稳定性试验结果(n=6)
2.5 药-时曲线与药代动力学参数
灌胃给予大鼠自制左氧氟沙星胃漂浮缓释微丸,其平均药-时曲线见图 2。以3P87软件计算药代动力学主要参数。结果半衰期(t1/2)为(7.51 ±1.04)h,药 -时曲线下面积(AUC0-t)为(119.73 ±11.34)μg/(mL·h),达峰时间(tmax)为(2.62 ±0.79)h,峰质量浓度(Cmax)为(7.91 ±1.45)μg/mL。
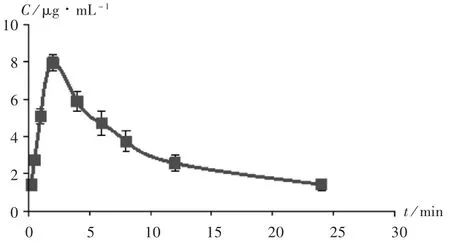
图2 平均药-时曲线(n=6)
3 讨论
在血样处理时,采用了加甲醇去除蛋白的方法,并与固液萃取法对比,发现均能较好地去除色图谱中内源性杂质对左氧氟沙星测定的干扰,但固液萃取操作较复杂[7],且成本更高,因此选择了前法;左氧氟沙星生物样品中药物质量浓度的测定方法采用了内标法,且选择结构相似的环丙沙星作为内标物,因环丙沙星具有相似的回收率,避免了处理过程中主药回收率偏低的问题。
目前,左氧氟沙星生物样品检测以高效液相色谱法居多[8-11],但本试验中选用了超高效液相色谱法,左氧氟沙星出峰快,流动相流速低,大大缩短了检测时间,减少了流动相的使用,提高了试验效率,节约了成本。另外,有文献采用超快速液相色谱-串联质谱法进行检测,虽也缩短了检测时间,但成本过高,操作烦琐,因此选用UPLC法测定更具有优势[12-13]。
该分析测定中配备了高灵敏度检测器,检测器流通池光程为60 mm,比普通检测器的流通池光程大6倍,灵敏度大大提高。选用了Waters ACQUITY UPLC BEH C18柱,左氧氟沙星色谱峰对称性好,峰宽较窄,柱效较高。本试验中还考察了不同流动相比例对左氧氟沙星质量浓度检测的影响,当流动相中磷酸二氢钾缓冲液与乙腈的比例为75∶25(V/V)时,分离情况最好。
综上所述,本研究中建立的UPLC法简单、快速、灵敏、准确,适用于大鼠血浆中左氧氟沙星的药代动力学研究。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
大众健康(2022年4期)2022-04-27
日用电器(2022年3期)2022-04-14
浙江化工(2022年1期)2022-02-19
中国土壤与肥料(2021年5期)2021-12-02
口腔护理用品工业(2021年4期)2021-11-02
中华养生保健(2020年10期)2021-01-18
今日农业(2020年22期)2020-12-14
中华养生保健(2020年3期)2020-11-16
中国科技纵横(2019年23期)2019-02-14
