
EDTA滴定法测定分银渣中的铅含量
2019-07-06 07:39:20范丽新
中国无机分析化学 2019年3期
郝 璐 范丽新
(1.北矿检测技术有限公司,北京102628;2.金属矿产资源评价与分析检测北京市重点实验室,北京102628)
前言
分银渣是阳极泥在分金、分银等步骤处理后所留下来的成分复杂的残渣,其中含有铅、铜、锑、铋、锌、锡、金、银等有价金属元素[1]。在矿产资源日益紧张的今天,分银渣作为一种二次资源,其经济价值和回收价值越来越高[2]。分银渣的成分复杂,锑、铋等杂质含量高,而现有的铅含量测定方法流程复杂且难以有效去除这些杂质。因此,亟需建立一种适用于测定分银渣中铅含量的分析方法。
目前测定矿产品中高含量的铅,一般采用硝酸或盐酸-硝酸体系溶解样品,在硫酸介质中形成硫酸铅沉淀进而通过过滤与其它元素分离,之后采用EDTA滴定法测定铅含量[3]。但在这个过程中分银渣中的锑、锡、铋等元素会发生水解,夹杂在硫酸铅沉淀中不易除去,影响铅含量的测定。以盐酸-硝酸-高氯酸体系溶解样品,并在高氯酸体系下加入氢溴酸,使氢溴酸与分银渣中的锑、锡、砷等元素生成易挥发的溴化物而消除这些元素的干扰。在硫酸体系下形成硫酸铅沉淀,经分离后,在缓冲溶液体系下用巯基乙酸掩蔽剂消除铋的影响。
1 实验部分
1.1 试剂
氟化铵溶液(200 g/L)、酒石酸溶液(200 g/L)、甲基橙指示剂(0.5 g/L)、巯基乙酸溶液(1+99)。
铅标准溶液:称取5.000 0 g金属铅(wPb≥99.99%)置于250 mL烧杯中,加入100 mL硝酸(1+1),低温溶解完全,煮沸赶尽氮的氧化物,取下冷却,用水移入1 000 mL容量瓶中并稀至刻度,混匀。
乙酸-乙酸钠缓冲溶液(pH值为5.5~6.0):200 g三水合乙酸钠溶于水中,加18 mL冰乙酸,用水稀释至 1 000 mL,混匀。
EDTA标准滴定溶液:c(Na2EDTA)≈0.010 mol/L,称取3.7 g 乙二胺四乙酸二钠(C10H14N2O8Na2·2H2O),加水微热溶解,冷却至室温,移入10 L试剂瓶中,用水稀释至10 L,摇匀,放置3 d后标定。
EDTA标准滴定溶液的标定:移取15.00 mL铅标准溶液于250 mL烧杯中,加入10 mL硫酸,加热至冒三氧化硫浓白烟,取下冷却。用水冲洗表面皿及杯壁,加水至60 mL,加热煮至液体体积为30 mL。按实验方法进行。平行标定四份,所得结果保留四位有效数字,其极差值不大于0.010 mg/mL时,取其平均值,否则重新标定。随同标定做空白实验。
1.2 实验方法
称取0.20 g试样(精确至0.000 1 g)于250 mL烧杯中,加入1~2 mL氟化铵溶液(200 g/L),10 mL盐酸,盖上表面皿,于低温处加热3 min。冷却后加入5 mL硝酸,继续加热3~5 min。冷却后加入5 mL高氯酸,低温缓慢加热至冒浓高氯酸烟,取下冷却,加入10 mL氢溴酸,低温缓慢加热至冒浓高氯酸烟后取下冷却,加入10 mL硫酸,缓慢加热至冒三氧化硫浓白烟,取下冷却。用去离子水冲洗表面皿及杯壁,加去离子水至60 mL,加入3~5 mL酒石酸(200 g/L),加热煮至液体体积为30 mL,取下冷却至室温,放置1 h以上。用慢速定量滤纸过滤,用硫酸(2%)洗涤液洗涤烧杯和沉淀6~7次,水洗涤烧杯和沉淀2次。将滤纸连同沉淀一起移入原烧杯,加入50 mL pH值为5.5~6.0的乙酸-乙酸钠缓冲溶液,用电炉加热微沸10 min,充分搅拌,取下冷却,用水冲洗杯壁至100 mL。滴加2滴二甲酚橙指示剂,加入1.5 mL巯基乙酸溶液(1%),用Na2EDTA标准滴定溶液滴定至紫红色变为亮黄色,此时即为终点。随同试样进行空白实验。
2 结果与讨论
2.1 分银渣主要成分
实验选取4个分银渣样品,分别称取0.10 g试样于25 mL刚玉坩埚中,碱熔酸化后转移至100 mL容量瓶中并稀释10倍。使用电感耦合等离子体原子发射光谱(ICP-AES)半定量法测定样品中含有的主要共存元素及其含量,测定结果如表1所示。
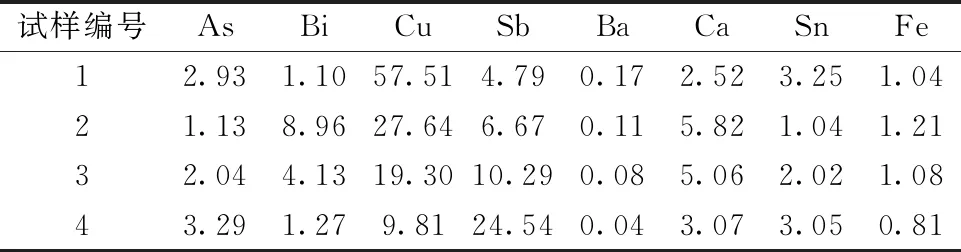
表1 分银渣主要共存元素及其含量
通过对ICP-AES的测定结果进行分析发现,分银渣中主要含有铜、锑、铋、砷、锡、钙、钡、铁等共存元素。然而,在EDTA法测定铅含量过程中,铜和锌等元素在生成硫酸铅沉淀和过滤的过程中能够与铅有效地分离,而其中少量夹杂的铁元素可以通过加入抗坏血酸进行掩蔽。此外,低于1%的钡和10%左右的钙含量对铅含量的测定无影响[3]。因此,用EDTA法测定分银渣的铅含量时需要消除砷、铋、锑、锡等主要共存元素的影响。
2.2 溶样方法
实验考察以下两种实验方法对杂质的去除效果。方法1:试样经氟化铵、盐酸、硝酸、硫酸和高氯酸共同分解后,在硫酸体系下加入10 mL氢溴酸,再加热至冒三氧化硫浓白烟,取下冷却。冷却至室温后加入酒石酸溶液和水煮沸。方法2:试样经氟化铵、盐酸、硝酸和高氯酸共同分解后,在高氯酸体系下加入10 mL氢溴酸,再加硫酸加热至冒三氧化硫浓白烟,取下冷却。冷却至室温后加入酒石酸溶液和水煮沸。按表2分别称取0.20 g分银渣试样(精确至0.000 1 g)置于250 mL烧杯中,然后采用以上两种方法对4个样品的铅含量进行测定,结果见表2。
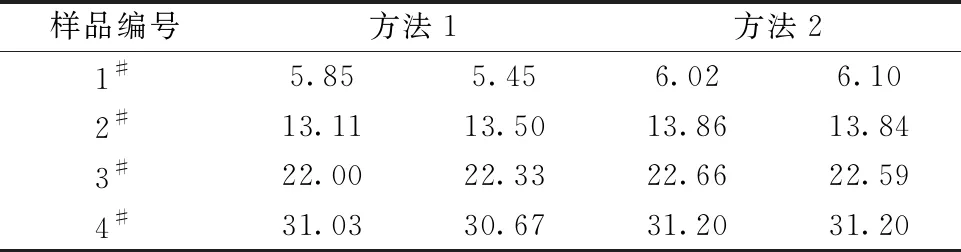
表2 溶样方式的选择
实验方法1溶样得到的结果普遍偏低,这可能是由于在硫酸体系下,铅已经形成硫酸铅沉淀,而锑、铋水解后形成的胶状物会吸附在硫酸铅沉淀中,此时加入氢溴酸没能充分和夹杂在沉淀中的杂质发生反应,残留的杂质影响硫酸铅沉淀的分离和洗涤效果。而方法2,在高氯酸体系下加入氢溴酸,氢溴酸能够充分和杂质反应生成溴化物,冒高氯酸烟时,形成的溴化物更容易随着高氯酸烟挥发除去。因此,实验选择方法2作为溶样方式。
2.3 锑干扰的消除
一般分银渣中锑的质量分数为0.1%~50%,而当锑的质量分数大于0.5%时,就会水解产生乳白色浑浊物,影响铅含量测定的准确度。当锑的质量分数大于2%时,由于浑浊物吸附杂质元素,影响铅元素的分离效果,使铅的测定结果偏低0.1%~0.2%[4]。按表3分别移取7份15 mL 铅标准溶液(5.000 0 g/L),向其中加入一定量的金属锑,测定结果如表3所示。
实验结果表明:当锑元素存在时会使铅的测定值明显偏低。在硫酸体系下加入氢溴酸,其未能充分与杂质反应,结果仍然偏低。而在高氯酸体系下加入氢溴酸一次除锑能够达到很好的效果。
2.4 铋干扰的消除
分银渣含有的铋会在硫酸冒烟过程中生成大量的硫酸铋,而实验过程中加入去离子水后,硫酸铋会部分水解产生不溶于水的Bi(OH)3·Bi(OH)SO4。该碱式盐和硫酸铅共沉淀,在滴定过程中铋会与Na2EDTA标准滴定溶液络合,从而对铅含量的测定造成正干扰[5]。
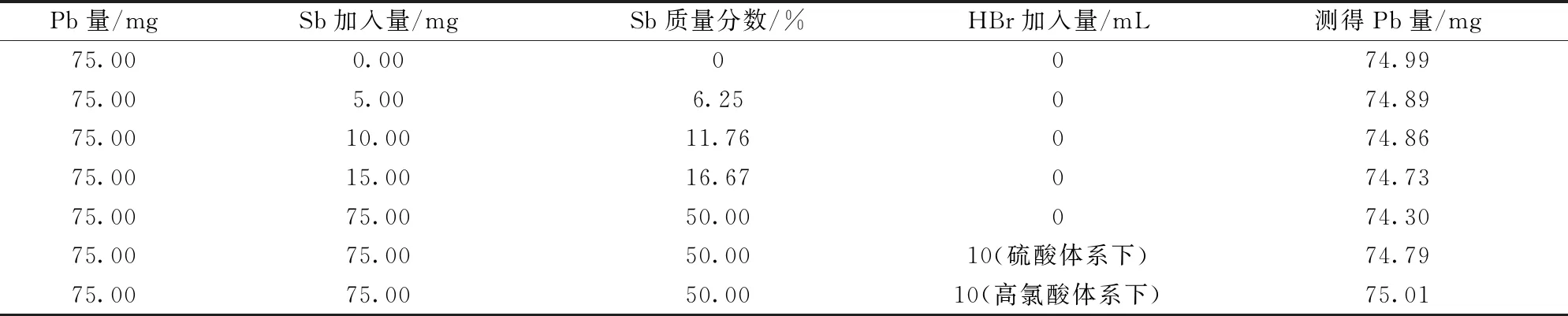
表3 锑的干扰及消除
按表4分别移取15 mL铅标准溶液(5.0000 g/L),向其中加入一定量的金属铋,按照实验方法进行溶样。同时考察滴定时加入不同量的巯基乙酸对测定结果的影响,测定结果见表4。
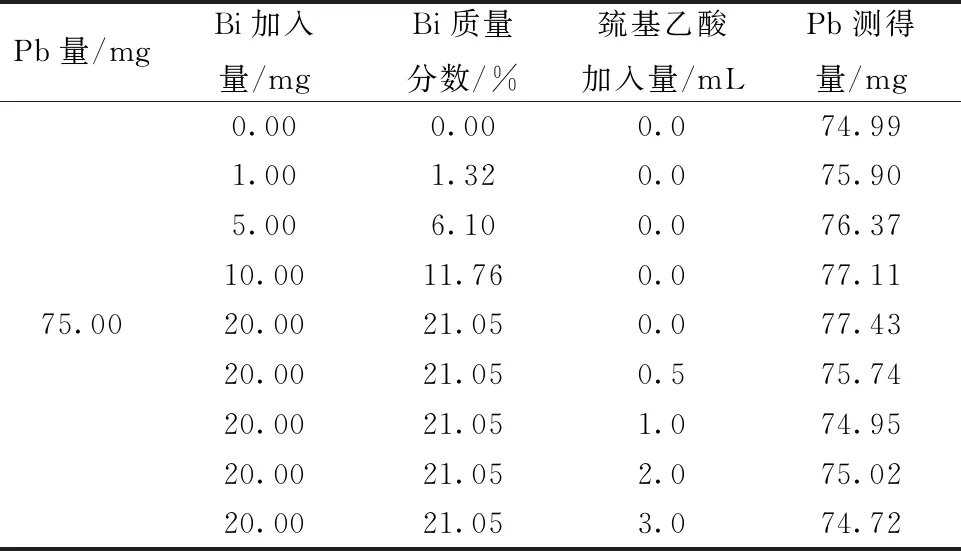
表4 铋的干扰实验
实验结果表明,当存在铋元素时会使铅的测定结果偏高。加入1.0~2.0 mL巯基乙酸,能够消除铋的干扰。而当巯基乙酸加入3.0 mL时,由于过多的巯基乙酸会络合铅,使得滴定终点不敏锐,导致测定结果偏低。因此在滴定时加入1.5 mL巯基乙酸溶液(1%),能够有效地消除铋对铅含量测定的影响。
2.5 陈化时间
硫酸铅的沉淀会直接影响铅与其它杂质元素的分离效果,因此选择合适的沉淀陈化时间以保证硫酸铅沉淀完全。分取四份15 mL 铅标准溶液(5.000 0 g/L)置于250 mL烧杯中,按实验步骤进行操作,沉淀后分别放置0.5、1.0、2.0、3.0 h,结果如表5所示。实验表明:当陈化时间为0.5 h时,测定结果明显偏低;陈化时间为1.0~3.0 h时,测定结果变化不大。因此实验选择将硫酸铅沉淀放置1.0 h以上。

表5 陈化时间实验
2.6 微沸时间
在将硫酸铅转化为乙酸铅的过程中,需要加热微沸以保证其转化完全。对合适的微沸时间进行考察,分取四份15 mL铅标准溶液(5.000 0 g/L),按实验步骤操作,在加入乙酸-乙酸钠缓冲溶液后,按表6选择不同的微沸时间进行实验。实验表明:当微沸时间小于10 min时,测得铅含量结果偏低,可能由于时间过短硫酸铅还未全部转化为乙酸铅;微沸时间在10~20 min时,测定结果变化不大。实验最终选择微沸10 min作为实验条件。

表6 微沸时间实验
2.7 杂质去除效果
按表7所示称取0.20 g(精确至0.000 1 g)的1#、2#、3#、4#试样,按照实验方法将试样经氟化铵、盐酸、硝酸和高氯酸共同分解后,在高氯酸体系下加入5 mL氢溴酸除去砷、锑、锡等元素,将溶液体积缩小至近干,转移至100 mL容量瓶中并稀释10倍,用ICP-AES测定其中的砷、锑、铋、锡等元素,并与原样品中的杂质含量进行比较,测定结果如表7所示。
实验表明,在高氯酸体系下加入氢溴酸能够有效去除砷、锑、锡等共存元素。与氢溴酸反应后,试样中砷和锡含量均低于0.1%,锑的含量低于0.5%,残留的少量锑可在后续步骤中与酒石酸反应进一步消除。
2.8 元素干扰实验
分取三份15 mL 铅标准溶液(5.000 0 g/L),按表8所示分别加入一定量的Cu、Sb、Bi、Sn、As,按实验步骤进行测定。从表8中的结果能够看出,该实验方法能够成功除去和分离上述元素,可以对分银渣中的铅含量进行准确的测定。
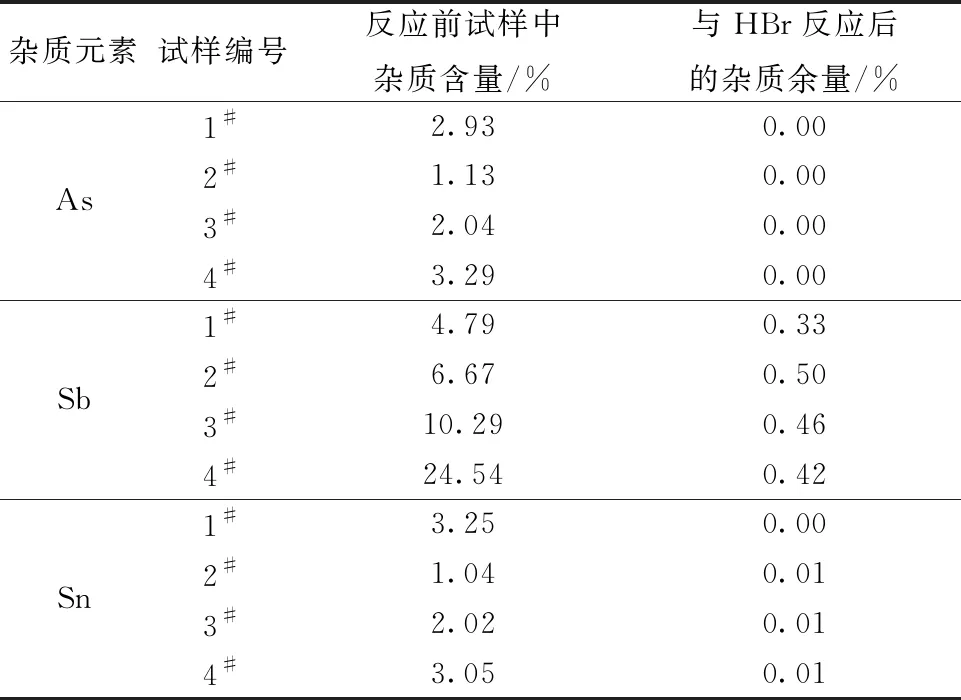
表7 反应前后样品中杂质含量

表8 综合元素干扰实验
2.9 精密度实验
对实验选取的4个分银渣样品独立进行11次测定,测定结果如表9所示。从表9中能够看出本方法测定分银渣中铅含量的相对标准偏差为0.32%~0.90%,能满足日常测定需求。
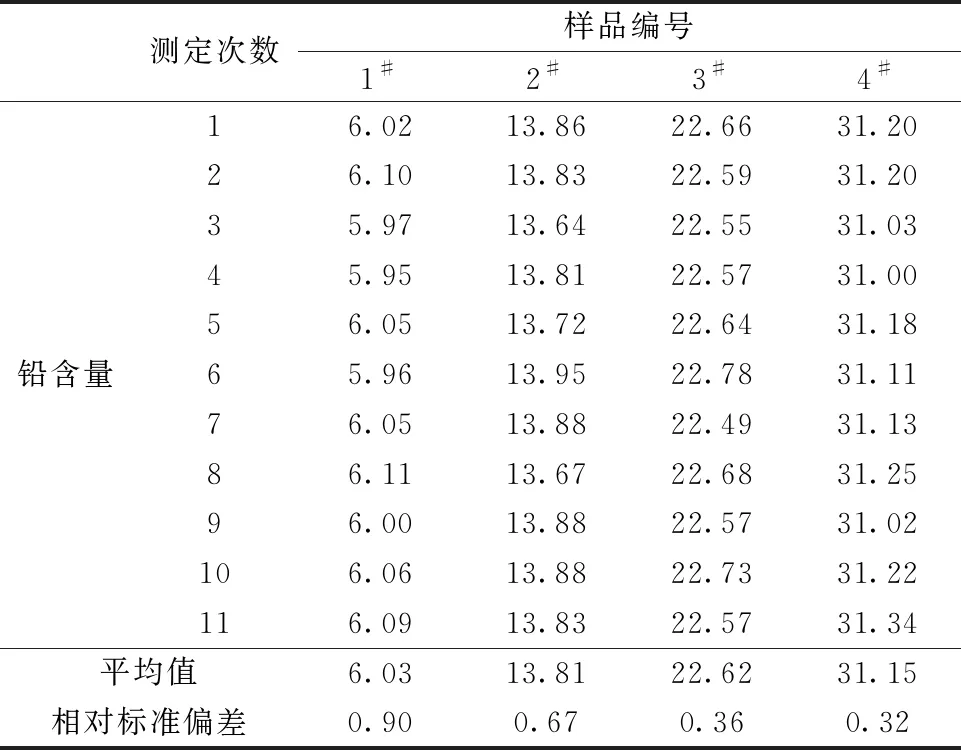
表9 精密度实验
2.10 加标回收实验
实验选取了2#、3#分银渣样品按实验方法进行铅的加标回收实验,结果如表10所示。从表中能够看出该方法的加标回收率为100%~102%,能够满足日常测定要求。
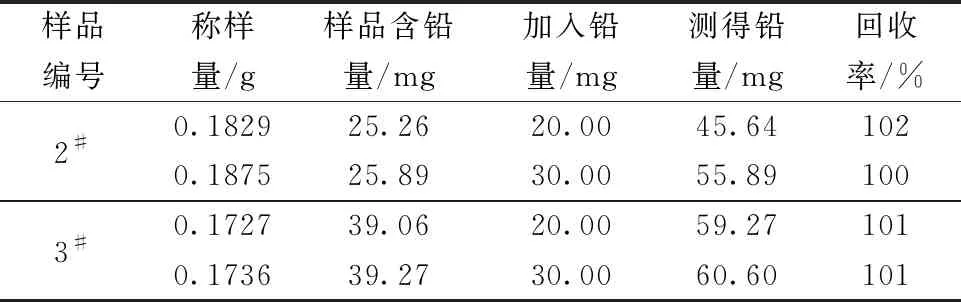
表10 加标回收实验
3 结论
实验表明,实验方法的相对标准偏差小于0.90%,加标回收率为100%~102%,适用于铅含量在6%~32%分银渣中铅含量的测定。方法快速、准确、便捷,满足日常生活中对分银渣中铅含量测定的要求。
猜你喜欢
云南化工(2022年8期)2022-08-16 09:38:30
中华养生保健(2020年5期)2020-11-16 01:44:30
有色金属科学与工程(2020年5期)2020-11-08 10:01:56
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年5期)2017-06-13 13:01:12
河北地质(2016年2期)2016-03-20 13:52:04
江西理工大学学报(2015年3期)2015-12-22 05:26:17
蓄电池(2015年3期)2015-07-02 03:22:45
应用化工(2014年12期)2014-08-16 13:10:46
无机化学学报(2014年6期)2014-02-28 17:32:05
