
DESI2基因干扰联合PI3K抑制剂对人胰腺癌细胞ASPC-1的影响
2019-06-24 08:35:50欧希张光涛徐喆陈景森谢勇刘吉奎刘晓平
肝胆胰外科杂志 2019年5期
欧希,张光涛,徐喆,陈景森,谢勇,刘吉奎,刘晓平
(1.北京大学深圳医院 肝胆胰外科,广东 深圳 518036;2.深圳市第三人民医院 肝病三区,广东 深圳518112;3.深圳市妇幼保健院 乳腺科,广东 深圳 518028)
细胞凋亡在肿瘤发病过程中(特别是在胰腺癌等恶性难治愈的肿瘤病变中)发挥着至关重要的作用[1],一旦细胞凋亡的调控机制异常或者凋亡受到阻断将会导致恶性肿瘤细胞的迁移与转化[2]。系列研究表明,肿瘤细胞中多条通路可以导致细胞异常激活,阻断细胞的正常凋亡过程,特别是AKT/mTOR信号通路,AKT蛋白中的T308和S473位发生磷酸化,会促进肿瘤的生长与分化,从而使肿瘤细胞表达失调,造成细胞过度增殖[3]。因此一些与AKT/mTOR信号通路相关的基因靶点药物正在研发中,但是胰腺癌作为一种消化道高病死率的癌症,单一的药物或者基因阻断不能很好地起到治疗效果。为了避免胰腺癌这种治疗抗拒,选择联合基因阻断可以为增强胰腺癌治疗效果提供新的途径。人凋亡相关新基因DESI2(desumoylating isopeptidase 2,被人熟知的名称是PNAS-4),是通过大规模测序获得的一种新基因[4],研究发现在肝癌和卵巢肿瘤中过表达DESI2基因都可以明显观察到肿瘤细胞的凋亡效应,肿瘤细胞活性明显受到抑制[5-6]。还有报道表明,DESI2基因与AKT/mTOR信号通路有相互作用[7],但是其作用机理暂无明确报道。因此在本实验中我们构建DESI2慢病毒干扰载体,并转入相应的胰腺癌细胞中,通过其与PI3K抑制剂联合作用,观察对胰腺癌细胞的生物学行为的影响及对胰腺癌细胞的杀伤效果。
1 材料和方法
1.1 细胞株及分组
ASPC-1细胞系购于上海中科院细胞库。将ASPC-1细胞分为四组:(1)溶剂对照组;(2)PI3K抑制剂组;(3)DESI2干扰+PI3K抑制剂组;(4)DESI2空载+PI3K抑制剂组。各组转染细胞使用Opti-MEM®培养基稀释DNA,制备DNA预混液,然后添加P3000TM试剂充分混匀,置于37 ℃、5% CO2孵箱中培养。
1.2 试剂与仪器
Lipofectamine®3000(18882752,Invitrogen);OPTI-MEM®I(331985-062,Gibco);Ultrapure RNA超纯RNA提取试剂盒(CW0581M,康为世纪);HiFiScript cDNA第一链合成试剂盒(CW2569M,康为世纪);Ul traSYBR Mixture(CW0957M,康为世纪);Fluo 4-AM(F312,Dojindo);超敏发光液(RJ239676,赛默飞);Mouse Monoclonal Anti-GAPDH(TA-08,中杉金桥,1/2000);Rabbit Polyclonal Anti-AKT(bs-2720R,Bioss,1/1500);Rabbit Polyclonal Anti-P-AKT(bs-0876R,Bioss,1/1500);Rabbit Monoclonal Anti-MTOR(ab32028,Abcam,1/1000);Rabbit Polyclonal Anti-P-MOTR(Ser2448,Cell Signalingr,1/1000);Rabbit Polyclonal Anti-PI3K(bs-2067R,Bioss,1/1500);Rabbit Polyclonal Anti-P-PI3K(Tyr458,Cell Signalingr,1/1000); Mouse Monoclonal Anti-Caspase3(bsm-33199M,Bioss,1/1000);Rabbit Polyclonal Anti-DESI2(bs-19976R,Bioss,1/1000);Annexin V-FITC/PI Apoptosis Kit(AP101-100-kit,MULTI SCIENCES联科生物);Cell Cycle Staining Kit(CCS012,MULTI SCIENCES联科生物);荧光细胞成像仪(ZOETM Bio-Rad);蛋白垂直电泳仪(DYY-6C,北京市六一仪器厂),超高灵敏度化学发光成像系统[Chemi DocTM XRS+,伯乐生命医学产品(上海)有限公司];荧光PCR仪[伯乐生命医学产品(上海)有限公司,CFX ConnectTM实时];NovoCyteTM流式细胞仪[NovoCyte 2060R,艾森生物(杭州)有限公司]。
1.3 DESI2干扰载体的构建
NCBI查找DESI2的基因,根据其基因的CDS序列设计shRNA靶序列,正义链引入BamHI酶切位点,反义链引入EcoRI酶切位点,退火形成双链,与载体pGreenpuro连接,构建慢病毒重组载体sh-DESI2-pGreenpuro。见表1。
1.4 细胞增殖检测
采用MTT法。将5×MTT用Dilution Buffer稀释成1×MTT,每孔加50 μL 1×MTT,在37 ℃孵育4 h,使MTT还原为甲臢,吸出上清液,每孔加150 μL DMSO使甲臢溶解,用酶标仪慢速振荡摇匀;酶标仪在550 nm波长处检测每孔的光密度。

表1 sh-DESI2序列
1.5 细胞周期及细胞凋亡检测
采用流式细胞仪。细胞转染48 h后,弃去培养液,PBS洗2次,加入胰蛋白酶消化1 min,加入含10%FBS的DMEM,将细胞吹打下来并吸取到1.50 mL离心管中,离心,弃去上清液,用PBS洗涤细胞两次(2 000 r/min离心5 min),收集1×105~5×105细胞,再加入1 mL 70%的乙醇溶液,放入4 ℃条件下2 h进行细胞固定。每管加PBS 1 mL,混匀,8 000 r/min离心3 min,弃上清。加入1 mL DNA staining solution,涡旋振荡5~10 s混匀。室温避光孵育30 min。上机检测,数据分析。
收集1×106~3×106个细胞,加1 mL PBS 1 500 r/min离心,3 min,洗两遍。用双蒸水将5×Binding Buffer稀释为1×Binding Buffer。取300 μL预冷的1×Binding Buffer重悬细胞。每管各加入3 μL Annexin V-FITC和5 μL PI-PE。轻微混匀后,室温避光孵育10 min。再向每管中加入200 μL预冷的1×Binding Buffer。混匀后上流式细胞仪检测。
1.6 细胞侵袭实验
细胞种板前1 d,血清饥饿12 h,常规消化、离心收集细胞,用低血清DMEM培养液(含0.2% FBS)混悬成密度为5×105/mL的单细胞悬液。将Transwell培养池放入24孔培养板中,上室内加入100 μL细胞悬液(约5×104细胞),下室内加入500 μL含10% FBS的DMEM培养液。置于5% CO2、37 ℃孵箱培养24 h后,PBS洗2次,用棉球小心擦去上室内细胞,4%多聚甲醛固定20~30 min,PBS洗2次,0.1%结晶紫染色30 min,PBS洗2次,去掉多余染料,显微镜下观察拍照。
1.7 qRT-PCR检测各基因的mRNA表达情况
取各组细胞进行RNA的提取,提取RNA后根据逆转录试剂盒合成cDNA,以cDNA为模板,在荧光定量PCR仪上进行检测,以GAPDH为内参,算出各组细胞中DESI2、AKT、Caspase3、mTOR和PI3K的相对表达量。引物序列见表2。

表2 引物序列
1.8 Western blotting检测各蛋白表达情况
取各组细胞加入组织裂解液,裂解30 min后,4 ℃、10 000 r/min离心10 min,小心吸取上清,即可获得总蛋白。根据BCA试剂盒测定蛋白浓度。蛋白变性后上样,再进行十二烷基苯磺酸钠凝胶电泳1~2 h,后湿法转膜30~50 min。4 ℃孵育一抗过夜;二抗溶液中室温孵育1~2 h。在膜上滴加ECL曝光液,在凝胶成像系统中曝光。
1.9 统计学分析
2 结果
2.1DESI2干扰载体的鉴定及效果验证
慢病毒重组载体转化感受态细胞,给予菌落PCR验证,理论上会得到629 bp大小的条带,由图1可知,电泳结果与理论值相符,均有阳性点。利用荧光定量PCR方法检测三条干扰序列中最具有干扰效果的序列,如图2A所示,三条干扰序列中sh-DESI2(3)具有干扰效果,因此选择sh-DESI2(3)进行以下实验。如图2B所示,与对照组相比,DESI2空载组的DESI2表达明显升高,DESI2干扰组则明显下降,差异具有统计学意义(P<0.05)。
2.2 各组细胞增殖情况
如图3所示,与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组,细胞的增殖受到抑制;与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组细胞活性明显升高。
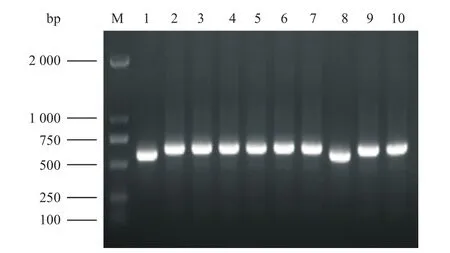
图1 菌落PCR电泳图
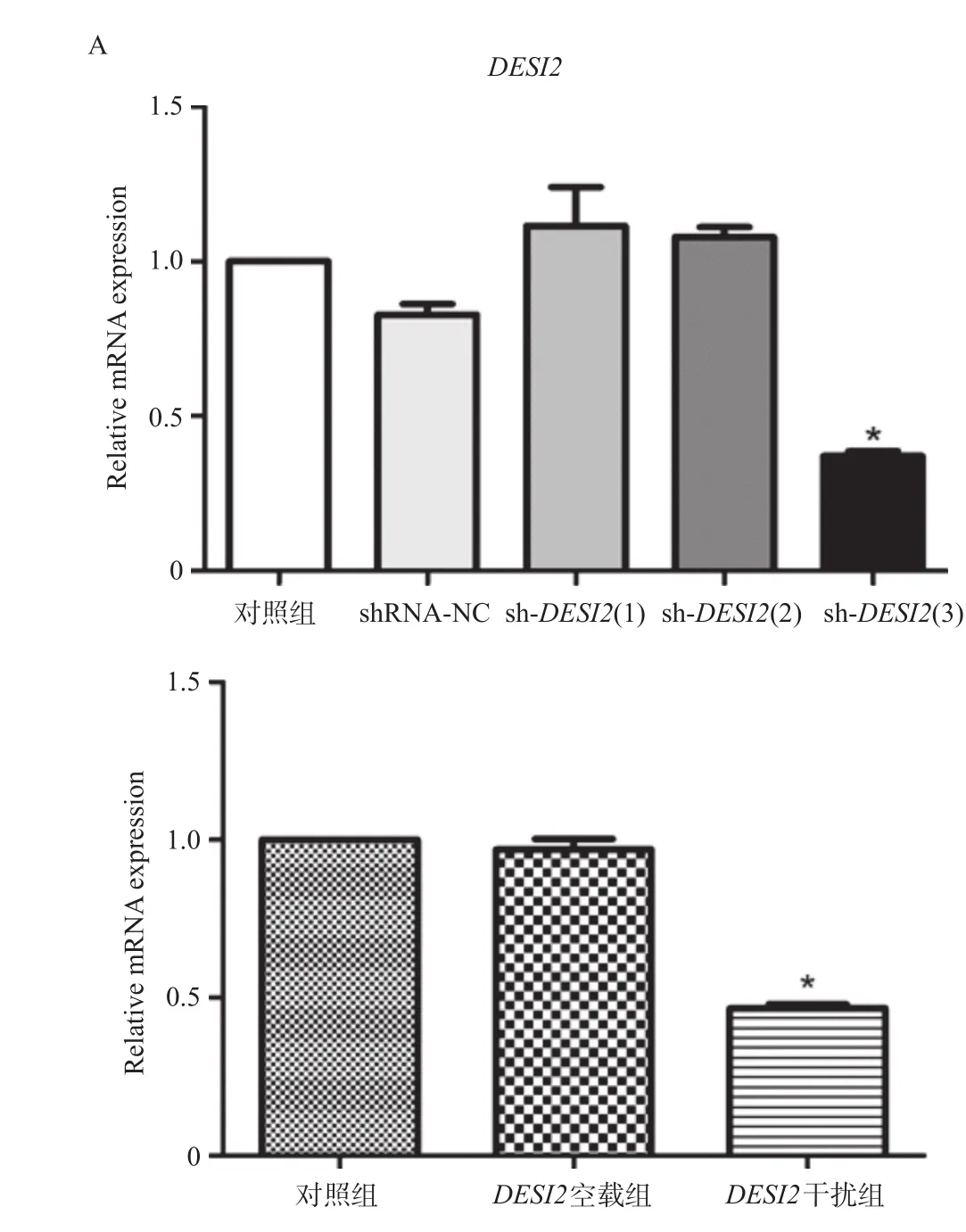
图2 DESI2干扰载体效果的验证
2.3 各组细胞周期变化情况
如图4所示,与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组细胞活性明显下降,G1期比例升高,S期和G2期比例降低。与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组细胞活性明显升高,G1期比例降低,S期比例升高。与对照组相比,PI3K抑制剂处理ASPC-1细胞可以抑制细胞增殖;而PI3K抑制剂处理DESI2干扰的ASPC-1细胞可以促进细胞增殖。
2.4 各组细胞凋亡情况
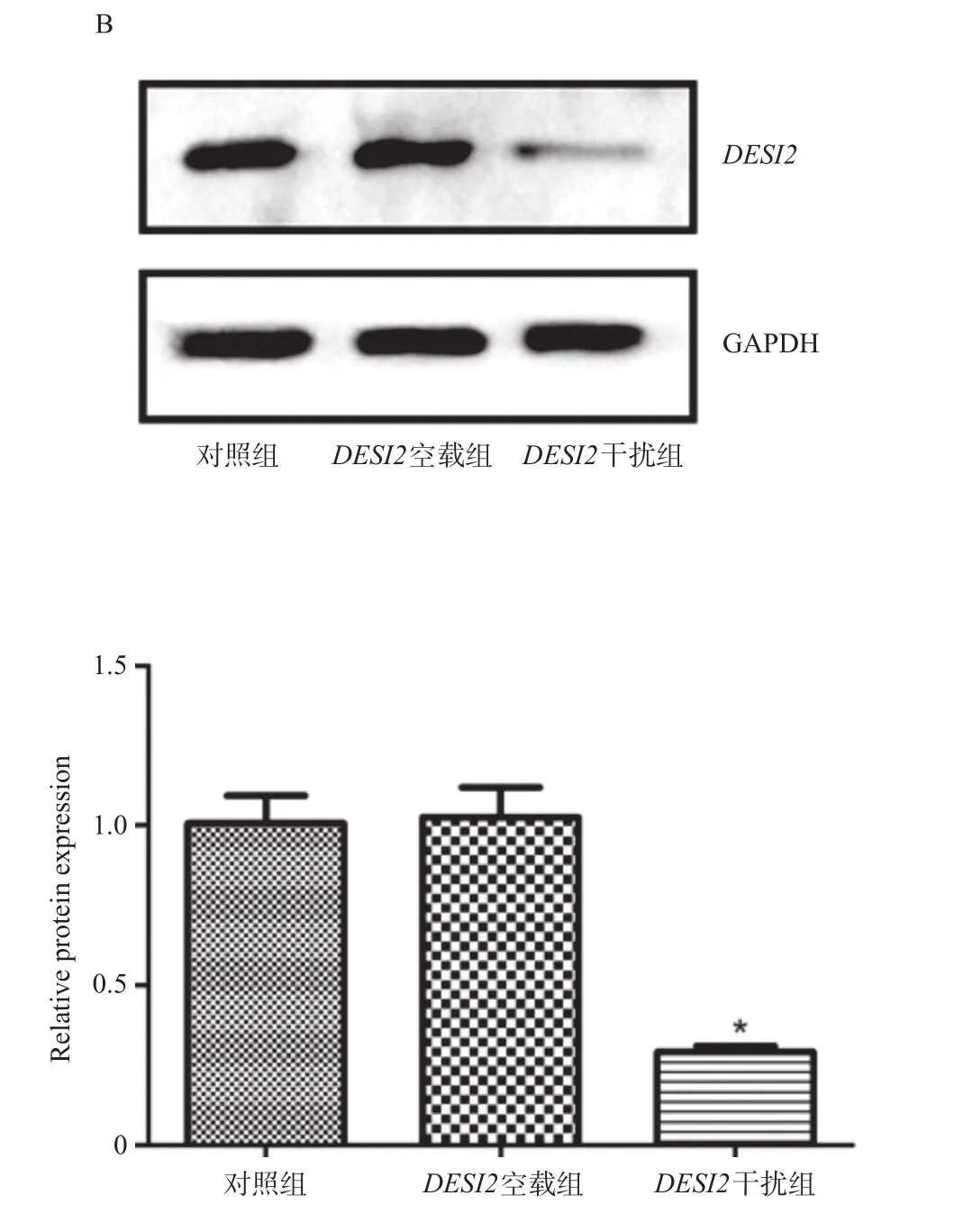
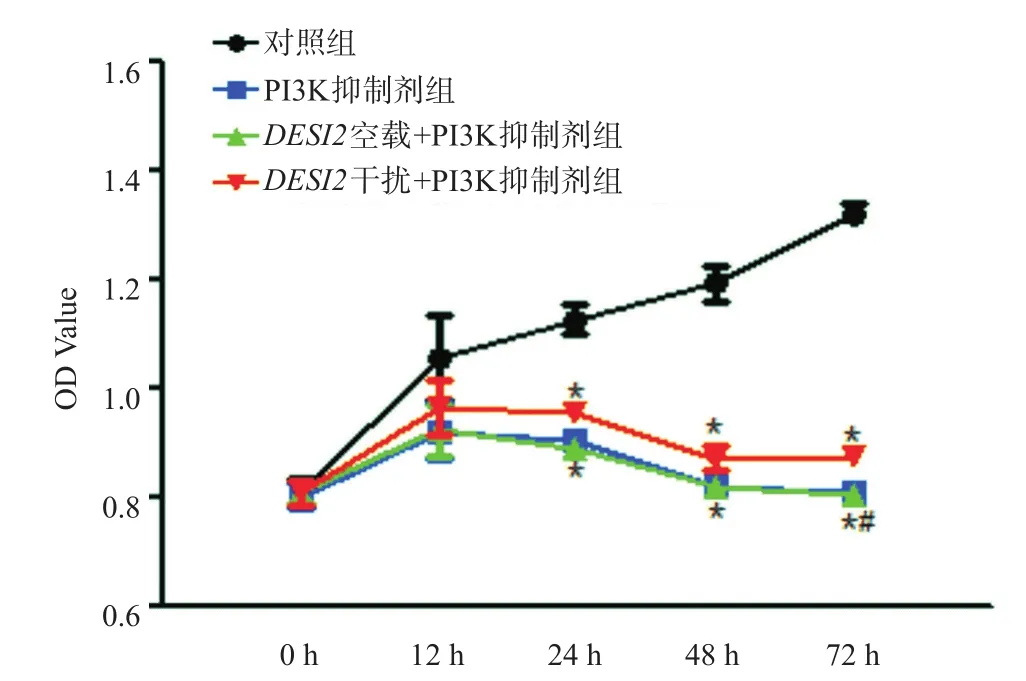
图3 MTT检测结果
如图5所示,与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组的凋亡明显升高;与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组细胞的凋亡明显下降,差异具有统计学意义(P<0.05)。
2.5 各组细胞侵袭情况
如图6所示,与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组的细胞侵袭明显下降;与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组细胞侵袭明显升高,差异具有统计学意义(P<0.05)。
2.6 各组细胞中的DESI2、AKT、PI3K、mTOR、Caspase3 mRNA和蛋白的表达结果以及p-AKT、p-PI3K、p-mTOR蛋白表达结果
与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组Caspase3 mRNA和蛋白表达升高,AKT、PI3K和mTOR及其磷酸化的蛋白表达量明显下降;与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组Caspase3 mRNA和蛋白表达下降,AKT、PI3K和mTOR及其磷酸化的蛋白表达量明显升高。见图7。
3 讨论
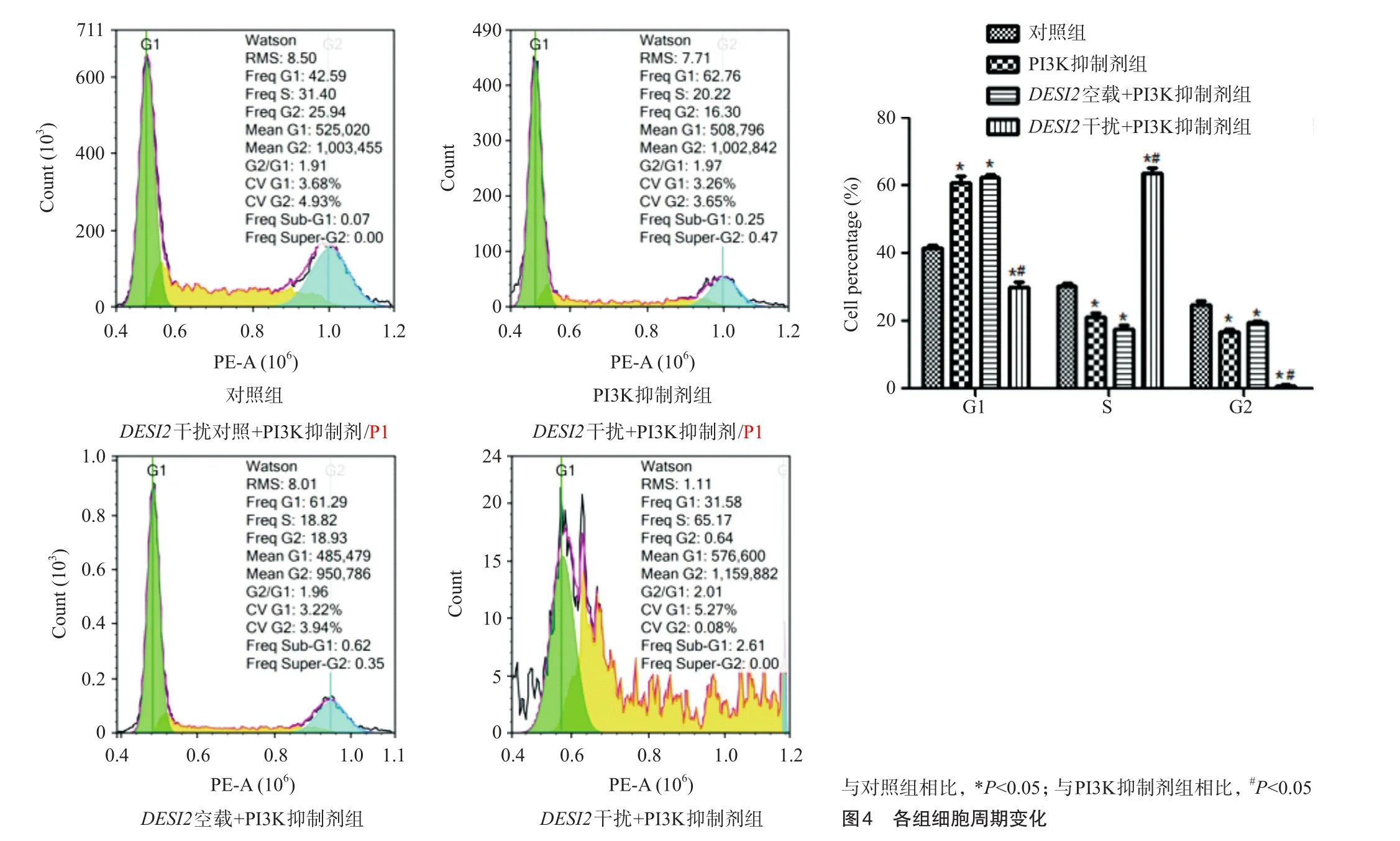
图4 各组细胞周期变化
基因靶向药物是肿瘤最新的治疗方式,靶点的选择和抗药性是其中的难点也是胰腺癌难以治疗和根治的主要原因之一[8]。如何解决药物的耐药性以及提高治疗的有效性是人们当前研究的热点。2002年美国国立卫生研究院在大规模测序中鉴定出一种新基因:DESI2基因,它定位于人染色体1q44,并且有5个外显子,可以编码1个由194个氨基酸残基组成的蛋白[9]。DESI2基因是一种促凋亡基因,有研究表明,DESI2不仅与机体生长发育相关,并且还参与了多种肿瘤的生物学行为[10]。有研究发现,DESI2基因在结直肠癌、肺癌等肿瘤中的表达均明显下降,可能DESI2基因在多种肿瘤中起着抑制肿瘤发生的作用[11-12]。AKT/mTOR是经典的信号通路之一,参与细胞生长、增殖、分化调节等各方面[13]。研究发现,活化的PI3K可以使AKT磷酸化而生成p-AKT,p-AKT进一步磷酸化其下游的酪氨酸和色氨酸残基,从而激活下游因子,抑制胰腺癌细胞的凋亡,促进细胞周期的运行,增加肿瘤血管形成和侵袭、转移的效果[14-17],因此通过采用PI3K抑制剂可以有效促进肿瘤细胞的凋亡通路的开启,其作用机理可能是通过调控肿瘤细胞中Caspase3蛋白的生成从而影响肿瘤细胞的凋亡过程[18],本研究与其结果类似,但是单一的抑制剂效果有限,因此需要寻找其相互作用的基因进一步来促进肿瘤细胞的凋亡以达到肿瘤治疗的效果和效率。
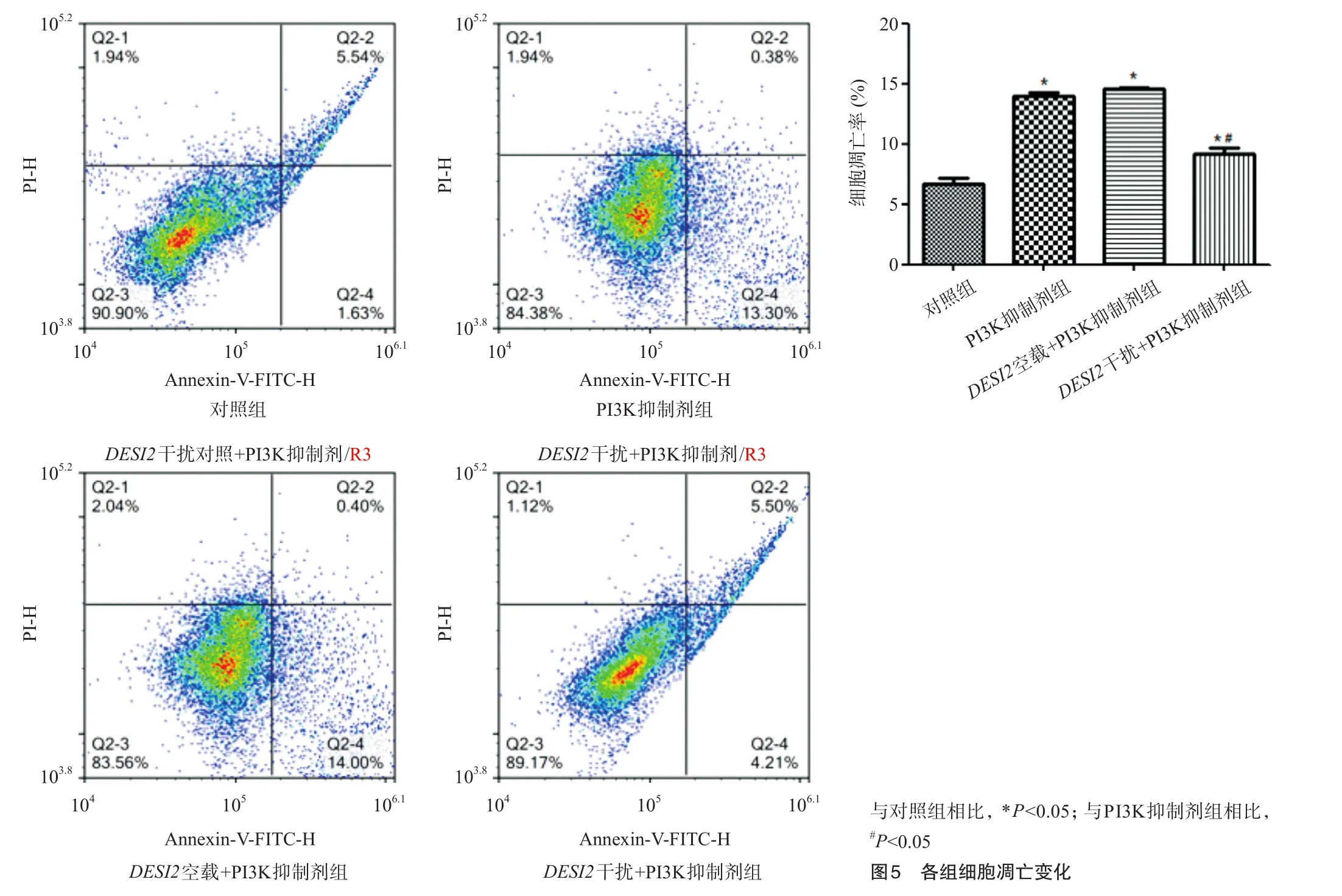
图5 各组细胞凋亡变化
在本实验中,与对照组相比,PI3K抑制剂组和DESI2空载+PI3K抑制剂组细胞活性明显下降,G1期比例升高,S期和G2期比例降低,凋亡明显升高,细胞侵袭能力明显下降,DESI2和Caspase3的表达明显升高,AKT、PI3K和mTOR及其磷酸化的表达量明显下降。与PI3K抑制剂组相比,DESI2干扰+PI3K抑制剂组细胞活性明显升高,G1期比例降低,S期比例升高,凋亡明显下降,细胞侵袭能力明显升高,DESI2和Caspase3的表达明显下降,AKT、PI3K和mTOR及其磷酸化的表达量明显升高。这表明PI3K/AKT/mTOR信号通路的功能状态能反馈调节上游DESI2基因的表达。得到这一结论的可能原因有以下几个方面:第一,单独的PI3K抑制剂可以明显达到抗肿瘤的效果,但是联合DESI2干扰后这一效果明显受到影响;第二,DESI2基因与PI3K/AKT/mTOR通路可以同时影响Caspase3蛋白的生成,因此两者之间可能有共同的凋亡途径,加入DESI2干扰后明显影响了肿瘤细胞的凋亡过程;第三,通过体外生物学实验表明,通过PI3K抑制剂处理后ASPC-1细胞的生存能力明显降低、侵袭能力明显减弱以及凋亡明显增加,但是加入DESI2干扰后这些效果明显受到影响;第四,通过蛋白表达水平检测发现,加入DESI2干扰后AKT、PI3K和mTOR及其磷酸化的表达量明显升高,而这些蛋白表达量的改变将进一步影响肿瘤细胞的凋亡过程。
总之,胰腺癌的细胞凋亡过程与AKT/mTOR及DESI2蛋白的表达密切相关,抑制DESI2蛋白的表达可能激活AKT/mTOR信号通路,促进肿瘤细胞的生长和浸润。因此本课题组下一步将继续对联合使用DESI2过表达载体与PI3K抑制剂进行研究,希望能为进一步提高胰腺肿瘤的治疗效果提供新的思路。
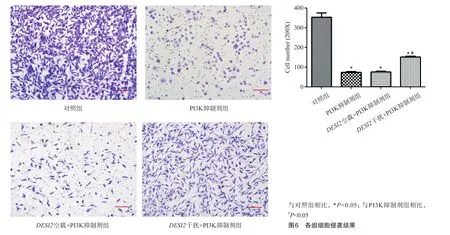
图6 各组细胞侵袭结果
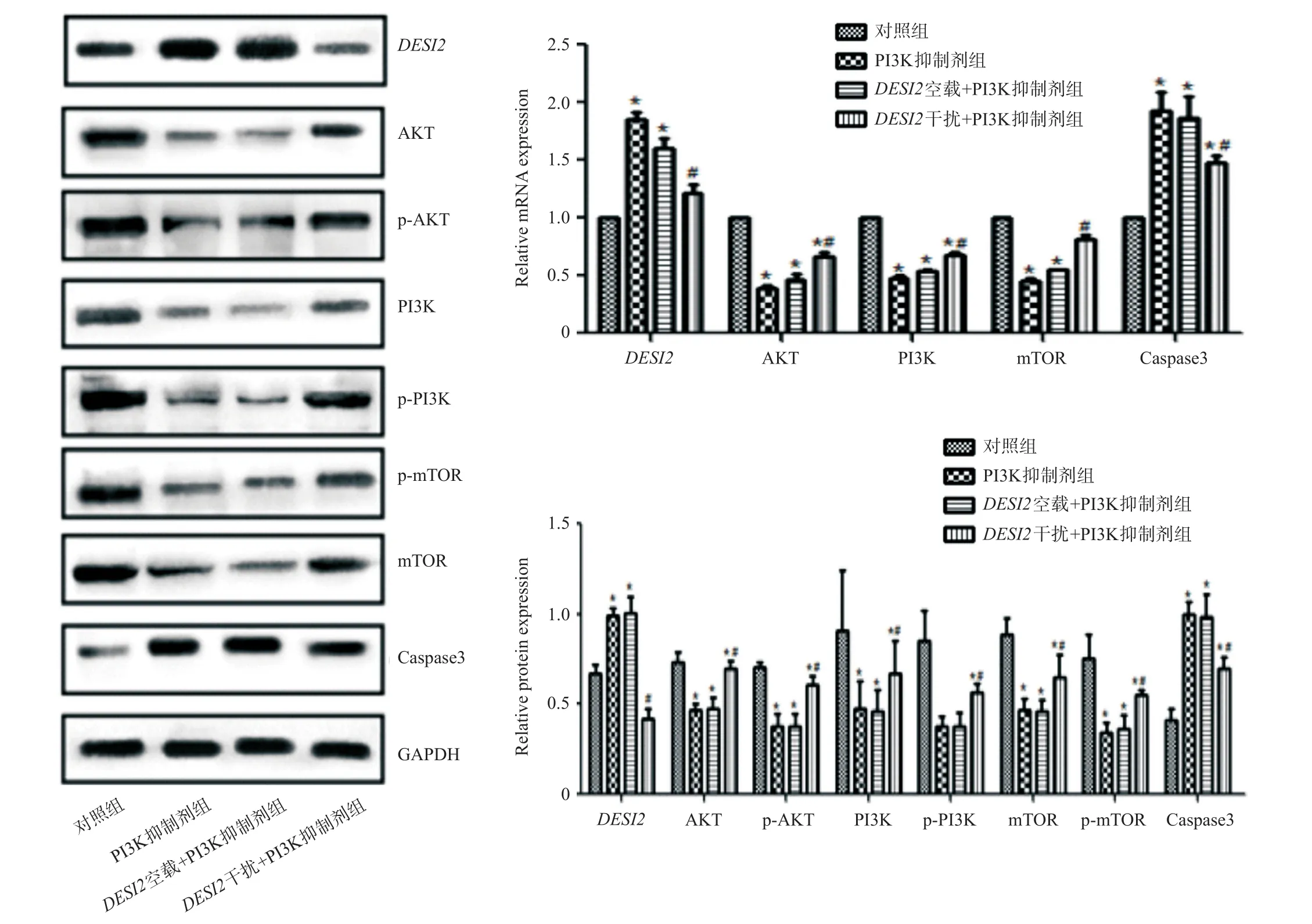
图7 各组细胞中的DESI2、AKT、p-AKT、PI3K、p-PI3K、p-mTOR、mTOR、Caspase3表达结果
猜你喜欢
保健医苑(2022年6期)2022-07-08 01:25:22
天津医科大学学报(2019年6期)2019-08-13 07:04:42
上海农业学报(2017年3期)2017-04-10 12:39:26
天津医药(2016年9期)2016-10-20 03:19:39
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
上海工运(2015年11期)2015-08-21 07:27:00
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
中国中医药现代远程教育(2014年22期)2014-03-01 04:33:08
遗传(2014年3期)2014-02-28 20:59:01
