
玉米叶绿素含量的全基因组关联分析
2019-06-22 07:48史大坤姚天茏刘楠楠邓敏段海洋王路林万炯高炯浩谢惠玲汤继华张雪海
中国农业科学 2019年11期
史大坤,姚天茏,刘楠楠,邓敏,段海洋,王路林,万炯,高炯浩,谢惠玲,汤继华,张雪海
玉米叶绿素含量的全基因组关联分析
史大坤1,姚天茏1,刘楠楠2,邓敏3,段海洋1,王路林1,万炯1,高炯浩1,谢惠玲1,汤继华1,张雪海1
(1河南农业大学农学院/省部共建小麦玉米作物学国家重点实验室,郑州 450002;2福建农林大学海峡联合研究院,福州 350002;3湖南农业大学农学院/湖南省玉米工程技术研究中心,长沙 410128)
【】叶绿素含量与作物产量呈正相关。通过提高叶绿素含量来提高作物产量是作物育种的方向之一。因此,利用全基因组关联分析(genome-wide association study, GWAS)解析玉米叶绿素含量的遗传基础,可为玉米高光效理想株型设计育种提供理论指导。以538份玉米自交系构成的关联群体为研究对象,在5个环境下,通过对其授粉后5 d的棒三叶(穗位叶、穗上叶、穗下叶)叶绿素含量进行测定,并借助覆盖玉米全基因组的558 629个单核苷酸多态性标记(SNPs),利用3种模型(Q、K和Q+K)对叶绿素含量进行全基因组关联分析,随后选择最优模型的GWAS结果并结合eQTL(expression quantitative trait loci)分析对叶绿素含量的自然变异进行解析。5个环境下,棒三叶叶绿素含量均遵从正态分布且叶绿素含量间呈正相关;方差分析表明棒三叶叶绿素含量的环境效应、基因型效应、基因型与环境互作效应均达到了极显著水平;此外,穗上叶、穗位叶和穗下叶叶绿素含量的遗传力分别为0.66、0.66和0.67。比较3种模型发现K模型对假阳性(I型错误)控制最好,在此模型下共检测到29个与棒三叶叶绿素含量显著关联的SNP(≤3.99×10-6),涉及到18个位点,共有76个候选基因落在这18个位点内,其中85.5%(65/76)的候选基因具有eQTL,11.8%(9/76)的候选基因与对应表型显著相关(<0.05),说明这9个基因可能是通过表达量变化来调控表型变异。在这76个基因中,60个候选基因有功能注释,功能涉及到能量代谢、物质输送代谢途径和生物合成调节等过程。此外还发现2个可以在不同环境或不同叶片共定位的位点,其中,共定位位点内的基因编码一种与AAE3高度相似的酰基活化酶,该基因通过提高α-酮戊二酸(ALA)和草酰乙酸含量进而影响氨基酸生物合成,提高籽粒赖氨酸含量,改善玉米品质。此外,ALA的合成会促使叶绿素含量升高,进而提高作物产量,推测该基因为最可能的候选基因。K模型对假阳性的控制效果最好,基于K模型,共检测到18个玉米叶绿素含量显著关联位点,发现多个参与叶绿素合成途径相关基因。
玉米;叶绿素含量;全基因组关联分析;高光合效率
0 引言
【研究意义】植物干物质的90%—95%通过光合作用产生,作物产量主要来源于叶片的光合产物[1]。而叶绿素含量是决定作物光合效率和生物量或产量的重要因素[2],也是高光效育种的重要育种目标之一。因此,对玉米叶绿素含量的遗传基础进行解析可为玉米高光效理想株型的设计育种提供理论支持。【前人研究进展】叶绿素是植物叶片的主要光合色素,也是植物光合作用的物质基础,已成为研究玉米生长特性及生理变化的重要指标[2]。孙红等[3]通过研究玉米生长期冠层反射光谱与叶绿素含量的关系,表明玉米拔节期和灌浆期冠层反射光谱与叶绿素含量呈正相关,从而认为叶绿素对玉米的生长至关重要。左宝玉等[4]研究表明玉米叶片的叶绿素含量为多基因控制的数量性状。文自翔等[5]通过亲本杂交建立的遗传群体结合连锁分析表明,QTL(quantitative trait locus)定位的准确性由亲本之间的遗传差异所决定,还发现一些位点在特定双亲间没有发生分离和重组,认为使用基于连锁分析的QTL定位存在一定的局限性。近年来,以连锁不平衡为基础的全基因组关联分析已被证明是一种解析复杂农艺性状遗传基础的强大工具[6-8]。随着测序技术发展、测序成本降低及统计模型改善,全基因组关联分析在作物QTL鉴定中被广泛应用[10]。Atwell等[11]用约200份拟南芥材料,结合216 130个SNPs标记,对其开花期、抗病及发育相关的107个表型进行GWAS,鉴定到大量主效位点,其中一些位点内含已知基因,说明GWAS对植物复杂性状遗传基础的解析是可行的。Thornsberry等[12]首次将关联分析用于玉米开花期研究,成功发掘到并验证其功能。而在玉米中,较早的GWAS则是对553份自交系成熟籽粒脂肪酸含量的分析[13]。在水稻和拟南芥中,部分叶绿素含量相关的基因已被克隆,WANG等[14]通过对水稻剑叶期的剑叶SPAD(soil and plant analyzer development)值、叶绿素a、叶绿素b进行测定,并结合高密度标记对不同亚型的水稻群体进行GWAS,检测到多个叶绿素相关性状显著位点,并发现已知基因也影响叶绿素含量;拟南芥编码二乙烯基叶绿素酯还原酶,该酶是二乙烯脱植基叶绿素a形成的关键酶,该基因突变会导致拟南芥叶片变黄[15]。在拟南芥发育初期,叶绿素合酶基因的突变体使叶片呈黄绿色,植株生长缓慢,野生型的绿色程度显著高于该突变体[16]。【本研究切入点】在玉米中,叶绿素含量QTL鲜有报道,且对其自然变异遗传基础的了解较少。玉米具有丰富的遗传多样性及快速的连锁不平衡衰减等特性,是进行全基因组关联分析的理想材料[17]。玉米不同叶位叶片的叶绿素含量决定各叶位叶光合强度,进而导致各叶位叶对籽粒产量贡献不同,而棒三叶(穗位叶及穗位叶的上下两片叶)作为有效光合层,对玉米籽粒产量贡献最大[18]。因此,通过全基因组关联分析对玉米棒三叶叶绿素含量的遗传结构进行解析,为快速获取叶绿素含量相关的标记及候选基因尤为重要,同时也可为高光合效率玉米品种的改良提供理论支持。【拟解决的关键问题】本研究以538份玉米自交系构成的关联群体为研究对象,通过对其棒三叶叶绿素含量进行测定,并结合高密度SNP标记,用3种不同的统计模型(Q、K、Q+K)对叶绿素含量进行GWAS分析,随后结合最优模型的GWAS结果及eQTL分析,探索玉米叶绿素含量的遗传基础,为玉米高光效的理想株型设计育种提供理论基础。
1 材料与方法
1.1 试验材料与田间种植
所用关联群体为华中农业大学严建兵教授提供,由538份玉米自交系构成。该群体于2012年夏季种植于河南鹤壁(12年鹤壁)鹤壁农业科学院浚县试验站(浚县,北纬N35°41′51.37″,东经E114°18′22.96″),2017年冬季种植于海南三亚(17年三亚)河南农业大学南繁基地(三亚,北纬N18°22′55.49″,东经E108°58′32.47″),2018年春季种植于湖南长沙(18年长沙)湖南农业大学试验基地(长沙,北纬N28°12′40.27″,东经E113°16′59.29″),2018年夏季种植于河南原阳(18年原阳)国家2011计划河南农业大学现代农业科技园区(原阳,北纬N35°03′58.63″,东经E113°56′2.78″)及河南永城(18年永城)棉花原种场(永城,北纬N33°52′53.11″,东经E116°27′11.51″)。采用单行区种植,单次重复,行长3 m,株距0.25 m,行距0.65 m,种植密度约63 000株/hm2。
1.2 叶绿素含量测定
为了准确测定玉米棒三叶的叶绿素含量,于授粉后5 d,使用手持SPAD仪(型号:SPAD-502 plus)对关联群体棒三叶(穗上叶(above the uppermost ear leaf)、穗位叶(uppermost ear leaf)、穗下叶(below the uppermost ear leaf))的SPAD值(叶绿素相对含量)进行测定。每行测定5株,对于每一株的棒三叶,选择每片叶的中间三分之一位置进行测定,重复3次(误差小于5%),3次测量均值作为该片叶的叶绿素含量;5株对应叶片的SPAD值的均值作为该自交系棒三叶对应叶片的叶绿素含量并用于后续的表型一般统计分析和全基因组关联分析。
1.3 数据处理与分析
利用Excel 2016对每个环境每个基因型棒三叶叶绿素含量的异常值进行剔除,并获得平均值。利用R语言的corrplot函数对不同环境下叶绿素含量进行相关性分析[19]。利用Excel 2016对5个环境下棒三叶叶绿素含量进行无重复双因素方差分析。利用R语言的lme4包[20]的混合线性模型[Y=(1|LINE)+(1|ENV)+ (1|LINE:ENV)]计算5个环境下每一个材料每一性状的最佳线性无偏预测(best linear unbiased prediction,Blup)值,基因型为随机因子,环境为固定因子,公式中,Y为性状数据,括号表示随机效应,“1|”表示分组,“:”表示互作,LINE表示所有材料,ENV表示环境。Blup值可减少不平衡数据造成的预测偏差,最终也用于一般统计分析及全基因组关联分析。广义遗传力的计算采用混合线性模型将基因型和环境作为随机效应,得到2和2的估计值。利用公式2=2/(2+2/)计算每个部位的叶绿素含量的遗传力,其中,2为遗传方差,2为协方差,为环境数[21]。
1.3.1 全基因组关联分析 所用基因型为Yang等[22]推算得到的覆盖玉米全基因组且最小等位基因频率≥0.05的558 629个SNP。考虑到很多标记间可能存在高度的连锁不平衡,利用GEC软件[23]计算该套标记的有效标记数(effective number,En),并以该有效标记数En作为判断性状-标记显著的依据(≤1/En)。统计学功效(即检测到真实显著关联的能力)是进行全基因组关联分析的首要考虑因素。因此,在Tassel 3.0软件[24]中使用3种模型,即只控制亲缘关系的K模型、只控制群体结构的Q模型、同时控制群体结构和亲缘关系的Q+K模型对关联群体棒三叶叶绿素含量分别进行全基因组关联分析,通过3种模型Quantile-quantile(QQ)图的比较,选择最优模型并对其结果进行进一步解析。
前期已有研究用558 629个SNP对该关联群体的连锁不平衡(linkage disequilibrium,LD)衰减程度进行评价,结果表明,该群体的LD衰减距离为50 kb(2=0.1)[25]。因此,定义一个显著SNP标记物理位置的上下游各50 kb的区间范围为一个位点。基于B73参考基因组(RefGen_v2),从玉米数据库MaizeGDB(http://www.maizegdb.org)中下载玉米全基因组基因列表并从列表中搜索每个显著位点内的所有候选基因。根据基因的功能注释及其在B73参考基因组中的表达部位及表达量,选择一个最可能的候选基因作为该位点的候选基因。
1.3.2 表达量QTL分析 利用RNA-seq技术深度测序368份关联群体自交系授粉后15 d的籽粒,每个自交系有50%以上的基因表达且至少有10个reads可用,共获得了28 850个基因的表达量[26],通过eQTL定位鉴定调控基因表达的位点,方法同表型的全基因组关联分析。
2 结果
2.1 玉米棒三叶叶绿素含量表型数据分析
通过对该关联群体叶绿素含量进行分析,发现多个环境的穗位叶叶绿素含量均高于穗上叶和穗下叶。同时发现除鹤壁穗下叶叶绿素含量的偏度值绝对值大于1外,其余环境棒三叶叶绿素含量偏度值的绝对值均小于1(表1),表明不同叶片的叶绿素含量均遵从正态分布,为典型的数量性状。
2.2 玉米棒三叶叶绿素含量的相关性分析、方差分析及遗传力
通过对不同环境玉米棒三叶叶绿素含量的相关性分析,发现不同环境的棒三叶(穗上叶、穗位叶、穗下叶)叶绿素含量间均呈正相关(图1),并且发现各环境下棒三叶叶绿素含量间的相关系数,大多在0.6以上,推测棒三叶叶绿素的生物合成调控存在着相互协同促进作用。
方差分析表明关联群体棒三叶叶绿素含量的基因型效应、环境效应、基因型与环境互作效应均达极显著水平(表2),说明棒三叶不同叶片的叶绿素含量存在显著的遗传变异,且受环境影响较大;若在环境影响较大的不同环境间检测到相同的位点或基因,则说明这些位点或基因可以稳定遗传。此外,穗上叶、穗位叶和穗下叶的叶绿素含量的遗传力分别为0.66、0.66和0.67。

表1 关联群体不同环境下玉米棒三叶叶绿素含量的描述统计分析
sd.:STDEVP,标准差,计算基于给定的样本总体的标准偏差;Ske.:SKEW,偏度,用来体现相对平均值的不对称程度;Kur.:KURT,峰度,用来体现一组数据的峰值
sd.: STDEVP. Calculate the standard deviation based on the given sample population; Ske.: SKEW. The degree of asymmetry used to represent the relative mean; Kur.: KURT. A peak value used to represent a set of data

a、b、c、d、e、f分别表示2012年鹤壁、2017年三亚、2018年长沙、2018年永城、2018年原阳及Blup等6个环境。字母后面的数字1—3分别代表穗上叶、穗位叶、穗下叶。下同

表2 5个环境玉米棒三叶叶绿素含量的方差分析
E:环境;G:基因型;G×E:基因与环境互作。**:在0.01水平上差异显著
E: environment; G: genotype; G×E: genotype-by-environment interaction. **: Significant at the 0.01 level
2.3 玉米棒三叶叶绿素含量的全基因组关联分析
2.3.1 阈值确定 利用GEC软件计算558 629个SNPs的有效标记数(En),结果表明,En为250 345,软件建议的3.99×10-6(1/En),该值作为关联分析的显著阈值。
2.3.2 关联分析结果 对6个环境下的棒三叶叶绿素含量的GWAS结果进行分析,通过比较3种模型的QQ图(图2),针对不同模型对假阳性的控制来看,Q模型(图中红点)最差,K模型(图中黑点)和Q+K模型(图中蓝点)优于Q模型。从对假阴性的控制效果看,Q+K模型比K模型稍微过于严格。因此,选用表现相对好的K模型的GWAS结果用于后续的位点挖掘和基因预测。基于K模型,在≤3.99×10-6下,6个环境一共检测到29个SNPs与棒三叶叶绿素含量显著关联,涉及到18个位点(图3;表3)。
对其他几个环境来说,在鹤壁,共鉴定到7个显著SNP,涉及到6个位点,分别是位于第8染色体与穗上叶叶绿素有关的1个位点,解释5.55%的表型变异;位于第5和第10染色体与穗位叶叶绿素有关的3个位点,分别解释5.36%、4.76%和4.75%的表型变异;其余2个与穗位叶叶绿素有关,位于第3和第7染色体,可分别解释6.52%和6.37%的表型变异。
在三亚,共检测到7个显著SNP(4个位点),其中1个位于第7染色体与穗下叶叶绿素相关的位点,解释6.07%的表型变异;另外3个位于玉米第1和第9染色体,与穗位叶叶绿素有关,解释的表型变异分别为5.43%、6.01%和6.59%。
在长沙,检测到5个SNP-性状关联,这5个SNP落在3个位点内,其中位于第1和第10染色体的2个与穗位叶叶绿素显著相关的位点,能分别解释5.89%和6.71%的表型变异;位于第10染色体上与穗上叶叶绿素含量显著相关的位点,能解释5.80%的表型变异。
在5个环境综合(Blup)条件下,共检测到10个SNP,涉及到5个位点。其中有3个位点与穗上叶叶绿素含量显著相关,分别位于第3、第5和第9染色体,解释的表型变异分别为5.10%、4.80%和4.86%;1个位于玉米第8染色体与穗位叶叶绿素有关的位点,可解释4.70%的表型变异,此位点在鹤壁也被检测到,为后续的进一步研究奠定了基础;此外,还有1个位于第5染色体,与穗下叶叶绿素含量显著关联,可解释5.03%的表型变异。利用线性回归模型计算了穗上叶、穗位叶与穗下叶叶绿素含量检测到的位点的总表型变异,分别可解释18.5%、26.2%和11.4%的表型变异;但仍有部分变异未能被解释,由此造成的缺失遗传力(missing heritability)可能因为存在微效位点或上位性互作导致,当前GWAS方法难以检测。
鉴定的18个位点,其中10号与11号位点共定位于第8染色体1.40—1.50 Mb区间内,分别解释5.55%和4.70%的表型变异,该区间内共有2个基因(表3)。17号和18号位点共定位于第10染色体148.59—148.69 Mb区间,分别解释6.71%和5.08%的表型变异,该区间内共有6个基因(表3)。共定位结果表明,这两个位点受环境影响较小,可在不同环境下稳定遗传;此外,2号位点可同时被长沙环境下的穗上叶与穗位叶叶绿素含量检测到(表3)。遗憾的是,原阳和永城均未检测到显著位点(3.99×10-6)(图3和表3)。表达量QTL分析(eQTL)发现76个候选基因中,85.5%的基因具有eQTL(65/76),表型与表达量之间的相关性分析发现,11.8%(9/76)的基因与对应表型显著相关(<0.05),说明这些基因可能是通过表达量的变化来调控表型变异(数据未列出)。
2.3.3 候选基因确定 由于该群体的连锁不平衡衰减距离是50 kb(2=0.1),因此,在峰值SNP的上下游各50 kb,即100 kb范围内搜索候选基因。一共有76个候选基因落在这18个位点内,其中60个具有功能注释(表3),根据基因功能注释及其在B73参考基因组中的表达部位及表达量,选择一个最可能的候选基因作为该位点的候选基因(表3)。
3 讨论
统计功效是GWAS首要考虑因素,对玉米来说,受群体结构影响,不同性状对不同模型的敏感程度不同。Yang等[27]研究表明,使用最优统计模型,可增加GWAS的统计功效。此外,刘坤等[28]使用3种模型(Q、K、Q+K)对玉米株高(plant height,PH)、穗位高(ear height,EH)、EH/PH、总叶片数(leaf number,LN)、穗上叶片数(leaf number above ear,LNAN)、LNAN/LN等6个性状进行GWAS,发现Q模型对假阳性的控制不好,Q+K模型对假阳性的过于严格,而K模型表现最优;Zhao等[29]使用3种相同模型对玉米籽粒、穗轴、苞叶、茎杆、叶片等5个组织的砷含量进行GWAS,发现K和Q+K模型对假阳性的控制过于严格,最终选择表现较好的Q模型的结果进行解析。本研究中,Q模型的表现最差,K模型和Q+K模型相当,但Q+K模型过于严格(图2),因此,选择K模型结果对玉米棒三叶叶绿素含量的遗传基础进行解析。

图2 6个环境下棒三叶叶绿素含量3种模型(Q、K、Q+K)的QQ图

表3 玉米棒三叶叶绿素含量候选基因及功能注释
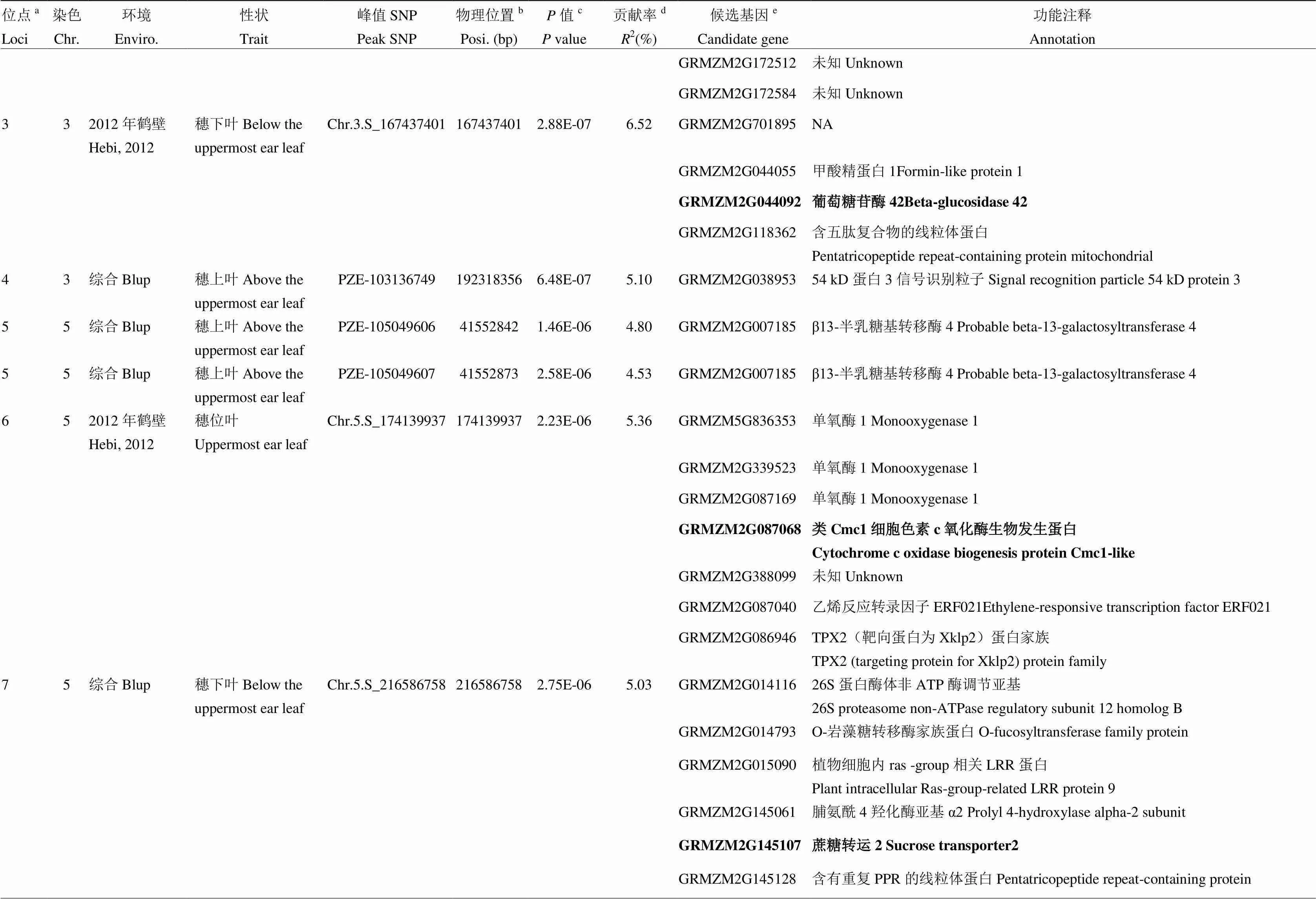
续表3 Continued table 3

续表3 Continued table 3
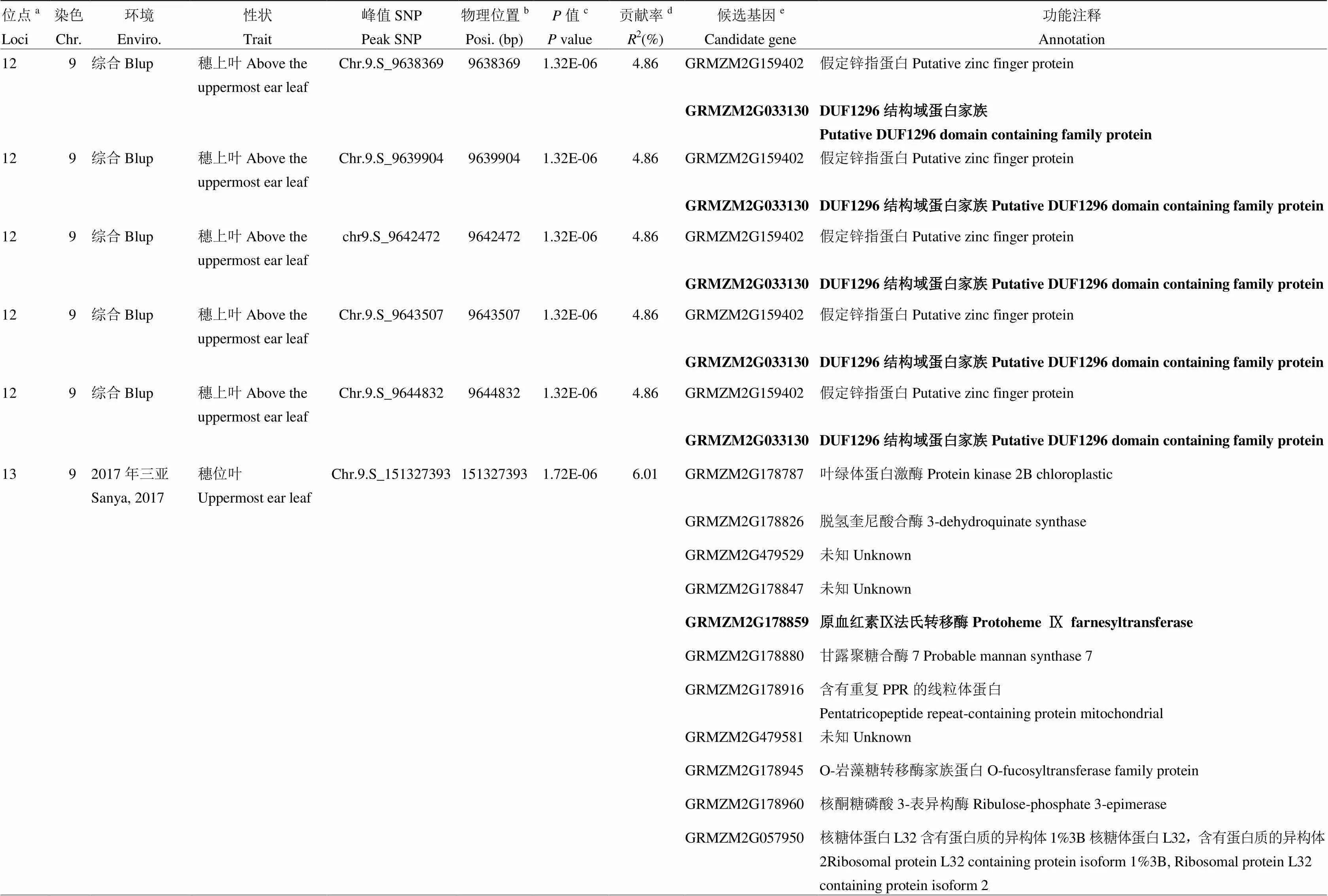
续表3 Continued table 3
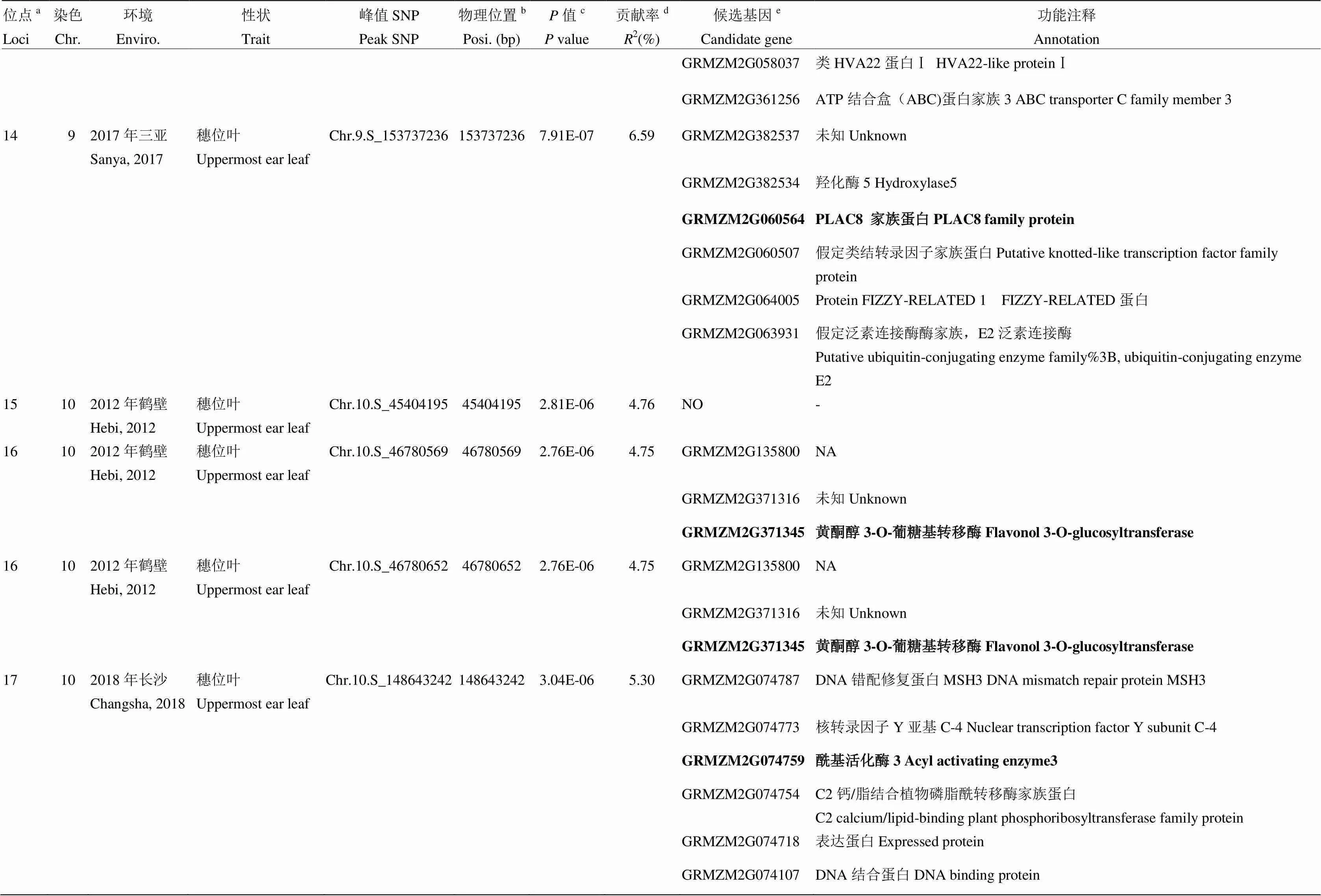
续表3 Continued table 3

续表3 Continued table 3
a所有有重叠区间的QTL被归为一个位点,位点的ID根据染色体及物理位置升序排序命名;b每个SNP的物理位置是基于B73参考基因组v2版本得到;c相应性状的值是通过K模型计算得到;d相应位点解释的表型变异;e位点内一个可能的生物学候选基因或离峰值SNP最近的候选基因。候选区间内最可能的候选基因用粗体标注
aAll QTLs with overlapping QTL regions were categorized as a loci, ID of loci was numbered according to chromosome and position by an ascending order method;bPhysical position of each SNP based on B73 RefGen_v2;cvalue of the corresponding trait calculated by K model;dThe phenotypic variance explained by the corresponding locus;eA plausible biological candidate gene in the locus or the nearest annotated gene to the lead SNP. The most possible candidate gene in each locus is marked in bold

图3 棒三叶叶绿素含量关联分析的曼哈顿图
赵永萍[30]通过对玉米单交种先玉335的棒三叶进行剪叶处理,探索玉米各部位叶片对百粒重、果穗干重、产量的影响,发现棒三叶的缺失会造成产量的明显下降,证明棒三叶对产量贡献至关重要。陈永欣等[31]对玉米棒三叶相关性状进行分析,得出棒三叶各叶片面积差异不大,在光合作用中发挥的作用近乎相同。本研究所测的棒三叶叶绿素含量相差不大,表明棒三叶在光合作用中可能发挥相似作用,与该结果吻合。唐海涛等[32]对65个玉米杂交种棒三叶的叶长、叶宽、叶肉厚度、叶面积、叶绿素含量、叶向值、光合速率、叶片干重和比叶重等性状进行分析,发现穗上叶、穗位叶、穗下叶间存在一定的相关性,且棒三叶比叶重(单位叶面积的叶片重量从小到大依次为穗位叶、穗上叶、穗下叶)和叶绿素含量(从小到大依次为穗上叶、穗位叶、穗下叶)呈现一定变化规律。左宝玉等[4]对玉米不同叶位叶片进行叶绿体超微结构观察及叶绿素含量进行测定,发现叶绿体的光合膜系统是叶绿素含量最高的部位,并且发现叶绿素含量和光合速率和产量之间呈显著正相关[33-34]。苏云松等[35]研究发现马铃薯叶片叶绿素含量(SPAD值)在正常日照条件下与马铃薯产量呈显著正相关,认为SPAD值的大小会直接影响马铃薯产量。王康等[36]研究表明夏玉米生育期的SPAD值与其产量呈正相关。以上研究均表明叶绿素含量对产量有重要贡献。
在前人研究中,拟南芥中发现的,将其进行过表达可显著抵抗氧化胁迫,可以修复突变体中受损的叶绿体膜结构,保护叶绿体膜,使叶绿素免受进一步降解[37]。在水稻中,与野生型相比,过表达植株的叶绿素a、叶绿素b和叶绿素总量分别比野生型增加5%、100%和25%,光合速率、生物量和产量相对增加20%、17%和16%[38]。作为的上位基因,表达的增加导致叶绿素含量显著增加,叶片叶色变为深绿色,光合速率和产量均显著提高[39]。在玉米中,也有叶绿素含量定位相关报道,王爱玉等[40]通过玉米A150-3-2和Mo17为亲本构建的189个F2单株为作图群体,分别在喇叭口期和开花期对叶绿素a,叶绿素b,叶绿素c和总叶绿素含量进行测定,共检测到32个与叶绿素含量相关的QTL。刘宗华等[41]以优良玉米杂交种农大108的203个F2:3家系为材料,利用复合区间作图法,在施氮和不施氮情况下,对玉米不同发育时期的叶绿素含量进行了QTL分析,认为叶绿素含量的QTL表达存在时空差异,其中2个QTL(和)可以在整个生育期被检测到,是生长发育必需的QTL。方永丰等[42]对已定位的玉米叶绿素QTL进行整合,利用荟萃分析(meta-analysis)发现多个共定位热区,并在这些热点区内挖掘与玉米叶绿素含量相关的候选基因。以上研究仅停留在连锁分析层面,需要构建作图群体,费时费力,且定位精度较低。随着测序技术的发展和测序成本的降低,关联分析在植物中被广泛使用,因其定位精度可达单基因水平,已成为挖掘植物基因的重要工具。
本研究对538份玉米自交系的棒三叶叶绿素含量(SPAD值)进行测定,结合55万个SNPs标记,通过全基因组关联分析,检测到29个与棒三叶叶绿素含量显著关联的SNP,涉及18个位点,共有76个候选基因落在这18个位点内,其中60个候选基因有功能注释,16个功能未知。已注释基因主要编码转运蛋白,结合蛋白,还原酶等,涉及功能包括能量代谢、生物合成调节及物质输送代谢等途径。18个位点中,有2个位点(第10和第11位点)在不同环境下共定位(表3),表明该位点受环境影响较小,在不同环境可稳定遗传。此外,另2个位点(第17和第18位点)在相同环境不同叶片被检测到(表3),说明这两个叶片的叶绿素含量有共同的遗传机制。此外还发现一些参与叶绿素合成及分解途径的基因。
3.1 共定位结果及候选基因分析
对定位结果进行分析发现2012年鹤壁穗上叶有关的第10位点和Blup条件下穗位叶相关的第11位点共定位,该位点内共有2个基因,分别为和。其中功能未知,而编码外被体β蛋白亚基(coatomer subunit beta),该基因仅在人类中有过描述,当敲除外被体β蛋白亚基时可使人类前列腺癌细胞凋亡速率升高,从而降低癌细胞的存活率[43]。但在植物中外被体β蛋白亚基的研究未见报道,因此,其作用机理尚不明确,推测其在玉米中也可能与细胞凋亡有关。
位于第10染色体上的与2018年长沙穗位叶相关的第17位点和2018年长沙穗上叶相关的第18位点共定位,位点内存在6个基因,(DNA mismatch repair protein MSH3)、(Nuclear transcription factor Y subunit C-4)、(acyl activating enzyme3,AAE3)、(C2 calcium/lipid-binding plantphosphoribosyltransferase family protein)、(Expressed protein)和(DNA binding protein)。编码一种与AAE3高度相似的酰基活化酶。的不透明表型即是由该酶减少所引起的α-玉米醇溶蛋白的浓度降低导致,通过氨基酸和代谢物的分析表明,该基因可能通过提高α-酮戊二酸(ALA)和草酰乙酸含量进而影响氨基酸生物合成,将该基因转入水稻中,同样会出现不透明胚乳表型。该基因的克隆为谷物种子贮藏蛋白含量的遗传调控提供了有效靶点[44]。也有研究表明,ALA的合成会促使叶绿素含量升高[45-47]。结合前人研究认为基因在参与调控玉米棒三叶叶绿素含量的同时也参与调控玉米籽粒赖氨酸的合成,当该基因过表达时叶绿素含量及籽粒中赖氨酸含量均升高。
3.2 叶绿素代谢相关候选基因
通过eQTL分析,发现2个参与叶绿素代谢的基因(和)。叶绿素属于四吡咯物质,当四吡咯物质不与蛋白结合时,会产生对植物有害的自由基。为了保护植物免受自由基的伤害,可以通过调节ALA的合成来阻止四吡咯物质的积累[45]。Pontoppidan等[46]证明谷氨酰-tRNA还原酶(Glu TR)在ALA合成中起拮抗作用,而血红素参与Glu TR活性的调节。在拟南芥中,可通过降低血红素加氧酶活性积累血红素,进而降低Glu TR活性[47]。本研究鉴定到的位于第9染色体的基因编码原血红素Ⅸ法氏转移酶,该酶可增加植物中血红素量积累,进而降低Glu TR活性,促进ALA的合成,从而促进叶绿素的合成。此外,7号染色体上检测到的编码NADPH-细胞色素P450还原酶2(CPR2)的基因参与叶绿素代谢。细胞色素P450(CYPs)是一种血红素结合蛋白,细胞色素P450接受电子后才能发挥活性。CPR2是细胞色素P450的主要电子供体,CPR2的存在使得细胞中血红素积累,谷氨酰-tRNA还原酶(Glu TR)活性降低,促使ALA合成,叶绿素合成增加[48]。并且,具有CPR功能,由于P450家族中的酶或基因功能多样,在玉米中并未明确其作用机理,仅在其他植物中有报道,表明CPR是一种将电子从NADPH转移到细胞色素P450酶的关键酶。在杉木中已明确CPR基因表达和酶活性的调控机制,通过Northern blot发现CPR在种子成熟前与萌发过程中的表达均受发育调控,而在种子和幼苗组织的子叶、胚根和大配子体中存在差异[49],推测其在玉米中具有类似功能,即CPR在玉米籽粒成熟前调控籽粒大小。
4 结论
基于K模型的GWAS结果,共检测到29个与棒三叶叶绿素含量显著关联的SNP,涉及到18个位点,其中2个位点可以在2个环境或2个组织中被共定位。18个位点内共有76个基因,其中85.5%的候选基因具有eQTL(65/76),11.8%的候选基因与对应表型显著相关(<0.05)。此外,共有76个候选基因落在这些位点内,其中60个基因具有功能注释并讨论了共定位以及与叶绿素代谢相关的基因。
致谢:本研究所用关联群体材料及基因型由华中农业大学作物遗传改良国家重点实验室严建兵教授提供,在此表示感谢。
[1] 刘红梅, 周新跃, 刘建丰, 邱颖波, 范峰峰, 徐庆国. 籼型杂交稻光合特性的配合力分析. 植物遗传资源学报, 2014, 15(4): 699-705.
LIU H M, ZHOU X Y, LIU J F, QIU Y B, FAN F F, XU Q G. Combining ability analysis of photosynthetic characteristics of indica hybrid rice., 2014, 15(4): 699-705. (in Chinese)
[2] 李合生. 现代植物生理学. 北京: 高等教育出版社, 2012.
LI H S.. Beijing: Higher education press, 2012. (in Chinese)
[3] 孙红, 李民赞, 张彦娥, 赵勇, 王海华. 玉米生长期叶片叶绿素含量检测研究. 光谱学与光谱分析, 2010, 30(9): 2488-2492.
SUN H, LI M Z, ZHANG Y E, ZHAO Y, WANG H H. Study of chlorophyll content in maize leaves during growing period., 2010, 30(9): 2488-2492. (in Chinese)
[4] 左宝玉, 李世仪, 匡廷云, 段续川. 玉米不同层次叶片叶绿体的超微结构和叶绿素含量变化. 作物学报, 1987, 13(3): 213-218.
ZUO B Y, LI S Y, KUANG T Y, DUAN X C. The changes of ulterastructure and chlorophyll content of chloroplast of leaves in different ranks in maize., 1987, 13(3): 213-218. (in Chinese)
[5] 文自翔, 赵团结, 郑永战, 刘顺湖, 王春娥, 王芳. 中国栽培和野生大豆农艺及品质性状与SSR标记的关联分析: Ⅱ.优异等位变异的发掘. 作物学报, 2008, 34(8): 1339-1349.
WEN Z X, ZHAO T J, ZHENG Y Z, LIU S H, WANG C E, WANG F. Association analysis of agronomic and quality traits with SSR markers inandin China: II. Exploration of elite alleles., 2008, 34(8): 1339-1349. (in Chinese)
[6] 李玮瑜, 张斌, 张嘉楠, 昌小平, 李润植, 景蕊莲. 利用关联分析发掘小麦自然群体旗叶叶绿素含量的优异等位变异. 作物学报, 2012, 38(6): 962-970.
LI W Y, ZHANG B, ZHANG J N, CHANG X P, LI R Z, JING R L. Exploring elite alleles for chlorophyll content of flag leaf in natural population of wheat by association analysis., 2012, 38(6): 962-970. (in Chinese)
[7] TIAN F, BADBURY P J, BROWN P J, HUNG H, BUCKLER E S. Genome-wide association study of leaf architecture in the maize nested association mapping population., 2011, 43(2): 159-162.
[8] ZHAO K, TUNG C W, EIZENGA G C, WRIGHT M H, ALI M L, PRICE A H. Genome-wide association mapping reveals a rich genetic architecture of complex traits in., 2011, 2: 467.
[10] 刘涛, 权文彦, 吴雪莲, 周露, 程宇坤, 姚方杰. 四川地方小麦品种产量与品质相关性状SSR标记位点的优异等位变异遗传解析. 麦类作物学报, 2015, 35(4): 449-456.
LIU T, QUAN W Y, WU X L, ZHOU L, CHENG Y K, YAO F J. Genetic analysis of SSR markers related to elite alleles of associated with yield and quality traits of sichuan wheat landraces., 2015, 35(4): 449-456. (in Chinese)
[11] ATWELL S, HUANG Y S, VILHJALMSSON B J, WILLEMS G, HORTON M, LI Y, MENG D, PLATT A, TARONE A M, HU T T, JIANG R, MULIYAFI N W, ZHANG X, ALNER M A, BAXTER I, BRAEHI B, CHORY J, DEARL C, DEBIEU M, DE MEAUX J, ECKER J R, FAURE N, KNISKERN J M, JONES J D, MICHAEL T, NEMRI A, ROUX F, SALT D E, TANG C, TODESCO M, TRAW M B, WEIGEL D, MARJORAM P, BOREVITZ J O, BERGELSON J, NORDBORG M. Genome-wide association study of 107 phenotypes ininbred lines., 2010, 465: 627-631.
[12] THORNSBERRY J M, GOODMAN M M, DOEBLEY J, KRESOVICH S, NIELSEN D, BUCKLER E S. Dwarf8 polymorphisms associate with variation in flowering time., 2001, 28(3): 286-289.
[13] ANDRÉ BELÓ, ZHENG P, LUCK S, SHEN B, MEYER D J, LI B, TINGEY S, RAFALSKI A. Whole genome scan detects an allelic variant offad2 associated with increased oleic acid levels in maize., 2008, 279(1): 1-10.
[14] WANG Q, XIE W, XING H, YAN J, MENG X, LI X, FU X, XU J, LIAN X, YU S, XING Y, WANG G. Genetic architecture of natural variation in rice chlorophyll content revealed by a genome-wide association study., 2015, 8(6): 946-957.
[15] NAGATA N, TANAKA R, SATOH S, Tanaka A. Identification of a vinyl reductase gene for chlorophyll synthesis inand implications for the evolution of Prochlorococcus species., 2005, 17(1): 233-240.
[16] WU Z, ZHANG X, HE B, DIAO L, WAN J. A chlorophyll-deficient rice mutant with impaired chlorophyllide esterification in chlorophyll biosynthesis., 2007, 145(1): 29-40.
[17] 陶士珩, 刘晓明, 储建华, 张荣梅, 杜丽萍, 罗泽伟. 混合群体连锁不平衡的衰减速率与基因定位. 科学通报, 2000, 45(21): 2274-2280.
TAO S H, LIU X M, CHU J H, ZHANG R M, DU L P, LUO Z W. Attenuation rate and gene location of linkagedisequilibrium in mixed population., 2000, 45(21): 2274-2280. (in Chinese)
[18] 赵可夫. 玉米抽雄后不同叶位叶对籽粒产量的影响及其光合性能. 作物学报, 1981, 7(4): 259-266.
ZHAO K F. Effect of the leaves of different positions in maize on the corn yield and the photosynthetic properties of those leaves after the growing out of the female flowers., 1981, 7(4): 259-266. (in Chinese)
[19] MCKENNA S, MEYER M, GREGG C, GERBER S. CorrPlot: An Interactive scatterplot for exploring correlation., 2015, 25(2): 445-463.
[20] CORETEAM R. R: a language and environment for statistical computing., 2015, 14: 12-21.
[21] XIAO Y, TONG H, YANG X, XU S, PAN Q, QIAO F, RAIHAN M S, LUO Y, LIU H, ZHANG X, YANG N, WANG X, DENG M, JIN M, ZHAO L, LUO X, ZHOU Y, LI X, LIU J, ZHAN W, LIU N, WANG H, CHEN G, CAI Y, XU G, WANG W, ZHENG D, YAN J. Genome- wide dissection of the maize ear genetic architecture using multiple populations., 2016, 210(3): 1095-1106.
[22] YANG N, LU Y, YANG X, HUANG J, ZHOU Y, ALI F, WEN W, LIU J, LI J, YAN J. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel., 2014, 10(9): e1004573.
[23] LI M X, YEUNG J M Y, CHERNY S S, SHAM P C. Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets., 2012, 131(5): 747-756.
[24] BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, BUCKLER E S. TASSEL: Software for association mapping of complex traits in diverse samples., 2007, 23(19): 2633-2635.
[25] LI H, PENG Z, YANG X, WANG W, FU J, WANG J, HAN Y, CHAI Y, GUO T, YANG N, LIU J, WARBURTON M L, CHENG Y, HAO X, ZHANG P, ZHAO J, LIU Y, WANG G, LI J, YAN J. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels., 2013, 45(1): 43-72.
[26] FU J, CHENG Y, LINGHU J, YANG X, KANG L, ZHANG Z, ZHANG J, HE C, DU X, PENG Z, WANG B, ZHAI L, DAI C, XU J, WANG W, LI X, ZHENG J, CHEN L, LUO L, LIU J,QIAN X, YAN J, WANG J, WANG G. RNA sequencing reveals the complex regulatory network in the maize kernel., 2013, 4: 2832.
[27] YANG X, YAN J, SHAH T, WARBURTON M L, LI Q, LI L, GAO Y, CHAI Y, FU Z, ZHOU Y, XU S, BAI G, MENG Y, ZHENG Y, LI J. Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection., 2010, 121(3): 417-431.
[28] 刘坤, 张雪海, 孙高阳, 闫鹏帅, 郭海平, 陈思远, 薛亚东, 郭占勇, 谢慧玲, 汤继华, 李卫华. 玉米株型相关性状的全基因组关联分析. 中国农业科学, 2018, 51(5): 821-834.
LIU K, ZHANG X H, SUN G Y, YAN P S, GUO H P, CHEN S Y, XUE Y D, GUO Z Y, XIE H L, TANG J H, LI W H. Genome-wide association studies of plant type traits in maize., 2018, 51(5): 821-834. (in Chinese)
[29] ZHAO Z, ZHANG H, FU Z, CHEN H, LIN Y, YAN P, LI W, XIE H, GUO Z, ZHANG X, TANG J. Genetic-based dissection of arsenic accumulation in maize using a genome-wide association analysis method., 2017, 91: 135-147.
[30] 赵永萍. 玉米不同叶位叶片与产量相关性研究. 现代农业科技, 2015, 644(6): 11-12.
Zhao Y P. Correlation between leaf position and yield of maize., 2015, 644(6): 11-12. (in Chinese)
[31] 陈永欣, 翟广谦, 李彦良, 王计虎. 糯玉米自交系、杂交种棒三叶与产量之间相关性分析. 玉米科学, 2001, 9(2): 50-52.
CHEN Y X, ZHAI G Q, LI Y L, WANG J H. Analysis on correlativity of three-ear-leaves of inbred line and hybrid-strain yield of glutinous maize., 2001, 9(2): 50-52. (in Chinese)
[32] 唐海涛, 张彪, 田玉秀, 余东梅, 陈洁, 康继伟. 玉米杂交种棒三叶光合性状比较研究. 玉米科学, 2009, 17(2): 86-90.
TANG H T, ZHANG B, TIAN Y X, YU D M, CHEN J, KANG J W. Comparison of photosynthetic characteristics of three ear-leaves hybrids maize., 2009, 17(2): 86-90. (in Chinese)
[33] 袁吉, 李艳玉, 蔚荣海. 鲜食糯玉米自交系叶绿素含量及其与产量的关系. 吉林农业, 2011(12): 64-65.
YUAN J, LI Y Y, WEI R H. Relationship of content of chlorophyll and yield of inbred line of fresh-eating waxy corn., 2011(12): 64-65. (in Chinese)
[34] 刘贞琦, 刘振业, 马达鹏, 曾淑芬. 水稻叶绿素含量及其与光合速率关系的研究. 作物学报, 1984, 10(1): 57-62.
LIU Z Q, LIU Z Y, MA D P, ZENG S F. A study on the relation between chlorophyll content and photosynthetic rate of rice., 1984, 10(1): 57-62. (in Chinese)
[35] 苏云松, 郭华春, 陈伊里. 马铃薯叶片SPAD值与叶绿素含量及产量的相关性研究. 西南农业学报, 2007, 20(4): 690-693.
SU Y S, GUO H C, CHEN Y L. Relationship between SPAD readings chlorophyll contents and yield of potato (L.)., 2007, 20(4): 690-693. (in Chinese)
[36] 王康, 沈荣开, 唐友生. 用叶绿素测值(SPAD)评估夏玉米氮素状况的实验研究. 灌溉排水学报, 2002, 21(4): 1-3.
WANG K, SHEN R K, TANG Y S. Evaluating nitrogen status with chlorophyll meter in summer corn., 2002, 21(4): 1-3. (in Chinese)
[37] ZHANG L, KUSABA M, TANAKA A, Sakamoto W. Protection of chloroplast membranes by VIPP1 rescues aberrant seedling development innyc1 mutant., 2016, 7(73): 533.
[38] WANG F, WANG G, LI X, HUANG J, ZHENG J. Heredity, physiology and mapping of a chlorophyll content gene of rice (L.)., 2008, 165(3): 324-330.
[39] HUANG J, QIN F, ZANG G, KANG Z, ZOU H, HU F, YUE C, LI X, WANG G. Mutation of OsDET1 increases chlorophyll content in rice., 2013, 210(210C): 241-249.
[40] 王爱玉, 张春庆. 玉米叶绿素含量的QTL定位. 遗传, 2008, 30(8): 1083-1091.
WANG A Y, ZHANG C Q. QTL mapping for chlorophyll content in maize., 2008, 30(8): 1083-1091. (in Chinese)
[41] 刘宗华, 谢惠玲, 王春丽, 田国伟, 卫晓轶, 胡彦民. 氮胁迫和非胁迫条件下玉米不同时期叶绿素含量的QTL分析. 植物营养与肥料学报, 2008, 14(5): 845-851.
LIU Z H, XIE H L, WANG C L, TIAN G W, WEI X Y, HU Y M. QTL analysis of chlorophyll content of maize under N-stress and no N-stress at different development stages., 2008, 14(5): 845-851. (in Chinese)
[42] 方永丰, 李永生, 白江平, 慕平, 孟亚雄, 张金林. 玉米持绿相关QTL整合图谱构建及一致性QTL区域内候选基因发掘. 草业学报, 2012(4): 175-185.
FANG Y F, LI Y S, BAI J P, MU P, MENG Y X, ZHANG J L. Construction of integration QTL map and identification of candidate genes for stay-green in maize., 2012(4): 175-185. (in Chinese)
[43] MI Y, SUN C, WEI B, SUN F, GUO Y, HU Q, DING W, ZHU L, XIA G. Coatomer subunit beta 2 (COPB2), identified by label-free quantitative proteomics, regulates cell proliferation and apoptosis in human prostate carcinoma cells., 2018, 49(1): 473-480.
[44] WANG G, SUN X, WANG G, WANG F, GAO Q, SUN X, TANG Y, CHANG C, LAI J, ZHU L, XU Z, SONG R. Opaque7 encodes an acyl-activating enzyme-like protein that affects storage protein synthesis in maize endosperm., 2011, 189(4): 1281-1295.
[45] MESKAUSKIENE R, NATER M, GOSLINGS D, KESSLER F, OP DEN CAMP R, APEL K. FLU: A negative regulator of chlorophyll biosynthesis in., 2001, 98(22): 12826-12831.
[46] PONTOPPIDAN B, GAMINIKANNANGARA C. Purification and partial characterisation of barley glutamyl-tRNAGlu reductase, the enzyme that directs glutamate to chlorophyll biosynthesis., 2010, 225(2): 529-537.
[47] GOSLINGS D, MESKAUSKIENE R, KIM C,LEE KP, NATER M, APEL K. Concurrent interactions of heme and FLU with Glu tRNA reductase (HEMA1), the target of metabolic feedback inhibition of tetrapyrrole biosynthesis, in dark- and light-grownplants., 2010, 40(6): 957-967.
[48] 贺丽虹, 赵淑娟, 胡之璧. 植物细胞色素P450基因与功能研究进展. 药物生物技术, 2008, 15(2): 142-147.
HE L H, ZHAO S J, HU Z B. Gene and function research progress of plant cytochrome P450s., 2008, 15(2): 142-147. (in chinese)
[49] TRANBARGER T J, FORWARD B S, MISRA S. Regulation of NADPH-cytochrome P450 reductase expressed during Douglas-fir germination and seedling development., 2000, 44(2): 141-153.
Genome-wide Association Study of Chlorophyll Content in Maize
SHI DaKun1, YAO TianLong1, LIU NanNan2, DENG Min3, DUAN HaiYang1, WANG LuLin1, WAN Jiong1,GAO JiongHao1, XIE HuiLing1, TANG JiHua1, ZHANG XueHai1
(1College of Agronomy, Henan Agricultural University/National Key Laboratory of Wheat and Maize Crop Science, Zhengzhou 450002;2Haixia Institute of Science and Technology, Fujian Agriculture and Forestry University, Fuzhou 350002;3College of Agronomy, Hunan Agricultural University/Maize Engineering Research Center of Hunan Province, Changsha 410128)
【】 Chlorophyll content was positively correlated with crop yield, improving crop yield by increasing chlorophyll content has become an important breeding goal in maize. Thus, elucidating the genetic basis of chlorophyll content using genome-wide association study (GWAS) can provide theoretical support for ideotype-based maize breeding with high photosynthetic efficiency. 【】 The association mapping panel (AMP) used in this study was consisted of 538 maize inbred lines, chlorophyll content of maize three leaves (above the uppermost ear leaf, uppermost ear leaf and below the uppermost ear leaf ) of the AMP was investigated at 5 days after pollination at five locations, then a GWAS with three models (Q, K, Q+K) were carried out using 558 629 single nucleotide polymorphisms (SNPs). The combination of optimal GWAS model with expression quantitative trait loci (eQTL) analysis, natural variation of chlorophyll content was further explored. 【】All traits measured at the five locations exhibited an approximately normal distribution and positive correlations between paired traits were also observed. Analysis of variance indicated that significant variations were observed across environment, genotype and the genotype × environment interaction. In addition, the heritability of chlorophyll content was 0.66, 0.66, and 0.67 for above the uppermost ear leaf, uppermost ear leaf and below the uppermost ear leaf, respectively. When test with the optimal GWAS model, K model has the greatest success in reducing false positive (type I errors) than other two models. Based on the result of K model, a total of 18 loci involving in 29 significantly SNP-traits associations were detected (≤3.99×10-6), and 76 candidate genes were found, including 42 genes that have functional annotation that involved in energy metabolism, biosynthetic regulation and material transportation and metabolic pathways. Of which, 85.5% (65/76) of the candidate genes have eQTLs and 11.8% (9/76) of the candidate genes were significantly associated with the corresponding phenotype (<0.05), indicating that these nine genes may affect phenotypic variation by regulating their expression. Moreover, two loci were found to be co-localized in two environments or leaves, the genewithin the co-localized locus, encodes an acyl-activating enzyme, highly similar to AAE3. It can increase the lysine content and improve maize quality by increasing the content of α-ketoglutarate (ALA) and oxaloacetate, in addition, ALA could promote chlorophyll biosynthesis and improve crop yield, this gene was considered as the most likely candidate gene. 【】The results indicated that K model having the best result in reducing the false positive. Based on the K model, a total of 18 loci associated with chlorophyll content and several candidate genes may be involved in chlorophyll synthesis pathway were identified.
maize (L);chlorophyll content; genome-wide association study; high photosynthetic efficiency
10.3864/j.issn.0578-1752.2019.11.001
2019-01-21;
2019-02-28
河南省科技攻关项目(182102110349)、省部共建小麦玉米作物学国家重点实验室自主设置课题(39990080)、河南农业大学博士科研启动金(30500563)、2019年度河南农业大学科技创新基金(KJCX2019A01)
史大坤,E-mail:912320091@qq.com。姚天茏,E-mail:yaotianlong0629@qq.com。史大坤和姚天茏为同等贡献作者。通信作者张雪海,Tel:0371-63558122;E-mail:xuehai85@126.com
(责任编辑 李莉)
猜你喜欢
军事文摘(2022年16期)2022-08-24
河北果树(2021年4期)2021-12-02
今日农业(2021年11期)2021-08-13
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2020-12-09
医药前沿(2020年20期)2020-11-10
阅读(科学探秘)(2020年8期)2020-11-06
中西医结合肝病杂志(2020年2期)2020-10-27
绿色科技(2019年2期)2019-05-21
河北农业科学(2019年6期)2019-03-21
