
妊娠 流产致APQ4抗体阴性的NMOSD复发1例报道
2019-06-21 02:24冯艳蓉
中国实用神经疾病杂志 2019年10期
刘 涛 陈 硕 冯艳蓉
东南大学医学院附属南京同仁医院神经内科,江苏 南京 211100
视神经脊髓炎谱系疾病(NMOSD)是一种主要累及视神经、脊髓和大脑特殊部位的中枢神经系统免疫相关性脱髓鞘疾病,临床上主要以视神经炎和长节段脊髓炎为主要特征[1]。NMOSD患者女性明显多于男性,发病年龄中位数为39岁[2-5],53.25%的国人NMOSD在妊娠或产后发病[6],考虑该病复发可能与妊娠关系密切,而且日益增加的证据表明APQ4抗体在发病机制中起着重要作用[7-9]。东南大学医学院附属南京同仁医院神经内科近期收治1例妊娠流产后致NMOSD复发,但血清及脑脊液APQ4抗体、MOG抗体均阴性的患者。
1 临床资料
1.1主诉患者 女,36岁,医生,主因“后枕部疼痛伴双手麻木14 d,加重伴双手无力2 d”入院。
1.2现病史患者于2017-04-10无明显诱因开始出现后枕部阵发性针刺样疼痛,以右侧明显,每次持续数秒钟,自觉在说话及行走时明显,伴双手中指、环指、小指麻木,后就诊我科门诊。查肌电图未见明显异常,予口服“甲钴胺片、维生素B1片、加巴喷丁”治疗,后枕部的疼痛较前明显好转,但逐渐出现双手无力,以右侧明显,右手不能持筷,右手环指及小指无法伸直,伴胸腹部(脐以上)麻木及双上肢麻木,于2017-04-23再次就诊我院门诊,门诊拟“脊髓炎?多发性硬化?”收住入院。病程中无双下肢麻木、无力,无言语不清,无饮水呛咳、吞咽困难,无嗝逆,无胸闷、呼吸困难,自发病以来饮食睡眠可,大小便正常,体质量无明显增减。
1.3既往史1 a前有双眼突发视力下降,当时我院眼科诊断“球后视神经炎”,查垂体MRI未见明显异常,予激素冲击治疗后双眼视力恢复如常。患者1个月前因出现阴道不规则流血至我院妇科就诊,诊断自然流产。
1.4月经史末次月经2017-02-07。
1.5体格检查T 36.5 ℃,P 74次/min,R 20次/min,BP 118/80 mmHg(1 mmHg=0.133 kPa),神清,两肺呼吸音清,未闻及干湿性啰音,心律齐,各瓣膜听诊区未闻及病理性杂音,腹软,肝脾未及,双下肢无水肿。专科检查:神清,精神可,言语流利,对答切题,双侧眼裂对称,双侧瞳孔等大等圆,直径3.0 mm,对光反射灵敏,双眼球活动自如,未见明显眼震。双侧额纹、鼻唇沟对称,伸舌不偏,悬雍垂居中,软腭上抬可,咽反射存在,双侧耳前区痛觉减退,粗测听力正常,气导>骨导,Weber居中,转颈、耸肩有力。全身肌肉无萎缩,颈屈肌肌力4级,伸肌肌力尚可,右手小指及环指伸肌肌力2级,余肢体肌力5级,肌张力正常,腱反射(+),右侧颈前C2~C4分布区痛觉减退,左手小指及小鱼际痛觉减退,右手小指及环指、小鱼际痛觉减退,双侧指鼻试验笨拙(右侧明显),双侧跟膝胫试验稳准,闭目难立征阴性,病理征未引出。颈抵抗,颈颏2横指。
1.6实验室检查血常规示WBC 6.9×109个/L,N 70.4%,HB 124 g/L,PLT 257×109个/L;急诊生化示空腹血糖6.20 mmol/L,余正常;甲功7项示ATPO 78 IU/mL;血脂常规、风湿三项、免疫八项、凝血功能、肿瘤7项未见明显异常;抗核抗体三项、抗中性粒细胞胞浆抗体均阴性;血清抗NMO抗体、抗MBP抗体、抗MOG抗体均阴性。
1.7脑脊液检查压力260 mmH2O(1 mmH2O=0.009 8 kPa);常规:白细胞数 5×106个/L,潘氏试验阴性,细胞总数870×106个/L;乳酸脱氢酶(LDH)+腺苷脱氨酶(ADA)正常;脑脊液生化(3项):脑脊液总蛋白0.42 g/L,葡萄糖3.63 mmol/L,氯121.4 mmol/L;脑脊液抗NMO抗体、抗MBP抗体、抗MOG抗体均阴性。
1.8影像学检查2017-04-23颅脑MRI平扫未见异常;颈椎磁共振示C5-6椎间盘膨出,颈椎退行性改变。延髓至C5椎体水平脊髓异常信号,性质待定(图1);颅脑MRI+颈椎MRI增强示延髓-颈髓5异常信号影,炎性病变?多发性硬化活动期?其他待排;胸椎MRI平扫未见明显异常;腰椎MRI平扫提示L1-2椎间盘轻度膨出,L2上缘许莫氏结节,L2椎体局部脂肪浸润,腰背部肌筋膜炎;2017-05-12颈椎MRI平扫示延髓-颈髓5异常信号影,较2017-04-23好转(图2)。
1.9诊断定位诊断:双侧耳前区痛觉减退,颈屈肌肌力4级,右侧颈前C2-4分布区痛觉减退,定位于颈髓C2-4水平;右手小指及环指伸肌肌力2级,左手小指及小鱼际痛觉减退,右手小指及环指、小鱼际痛觉减退,定位于颈髓C6-7水平,颈抵抗,颈颏2横指,定位脑膜。定性诊断:定性为脱髓鞘疾病。考虑患者1 a前有视神经炎史,治疗后未留后遗症。本次为延髓、颈髓病变,具有时间、空间多发特点,结合颅脑MRI影像学特点,诊断视神经脊髓炎谱系疾病。
1.10治疗入院后给予甲泼尼龙静滴1 g/d,连续3 d;500 mg/d,连续3 d;240 mg/d,连续3 d;120 mg/d,连续3 d;60 mg/d,连续3 d;后改泼尼松60 mg/d,晨顿服。病程第2天联用丙种球蛋白20 g/d静滴,连续5 d;同时辅以补钾、护胃、营养神经、改善微循环等治疗。住院治疗3周患者出院。出院后泼尼松逐渐减量至20 mg/d维持治疗,病程1个月左右再次丙种球蛋白20 g/d静滴,连续3 d。
1.11预后3个月后随访患者双上肢无力、麻木完全缓解,无后遗症,已参加工作。1 a后随访无复发。
2 讨论
WINGERCHUK等[10]于2007年将一组发病机制和临床特征与视神经脊髓炎(NMO)相似的中枢神经系统脱髓鞘疾病统一命名为视神经脊髓炎谱系疾病(NMOSD)。血清中水通道蛋白4(APQ4)抗体被认为是NMOSD相对特异的生物学标志物。2010年欧洲神经科学协会联盟(EFNS)根据AQP4抗体的表达,将NMOSD分为AQP4抗体阳性和APQ4抗体阴性的NMOSD[11-12]。2015年,由WINGERCHUK等[13]专家组重新修订了NMOSD统一的诊断标准。该诊断标准把NMOSD的临床表现分为6个核心症状,包括视神经炎、急性脊髓炎、极后区综合征、急性脑干综合征、间脑临床综合征、大脑综合征。按照血清APQ4-IgG抗体监测结果进行分类,包括APQ4抗体阳性、APQ4抗体阴性以及无法检测APQ4抗体的NMOSD。其中APQ4抗体阴性的NMOSD诊断标准:(1)1次或多次临床发作累及至少2项核心临床症状,并且满足如下所有要求:①其中至少1项是视神经炎、急性纵向延伸横贯性脊髓炎或最后区综合征;②至少2项核心症状临床特征空间上播散分布;③如果适用,满足额外的MRI要求。(2)APQ4抗体阴性(用最可靠的检测方法)。(3)排除其他诊断(核心临床症状:①视神经炎;②急性脊髓炎;③极后区综合征:其他原因难以解释的呃逆或恶心、呕吐;④急性脑干综合征;⑤具有NMOSD典型间脑MRI病灶的症状性嗜睡或急性间脑临床综合征;⑥具有NMOSD典型大脑病灶的症状性大脑综合征。APQ4抗体阴性的MRI要求:(1)急性视神经炎:须有脑MRI正常或仅有非特异性白质病灶,或视神经MRI T2高信号或T1强化病灶累及一半以上视神经或累及视交叉;(2)急性脊髓炎:须脊髓内病灶超过3个椎体节段或具有急性脊髓炎史的患者存在连续3个节段的脊髓萎缩;(3)最后区综合征:须有延髓背侧/最后区病灶;(4)急性脑干综合征:须有室管膜周围脑干病灶)。该患者系青年女性,1 a前有视神经炎病史,经治疗未遗留后遗症。本次为自然流产后10 d余出现后枕部疼痛及双手麻木、无力的急性脊髓炎症状,颅脑MRI示延髓、颈髓病灶超过3个椎体的影像学特点,实验室检查血清及脑脊液APQ4抗体阴性,具有时间、空间多发的临床特点,符合WINGERCHUK等专家组重新修订的APQ4抗体阴性的NMOSD诊断标准,故诊断明确。
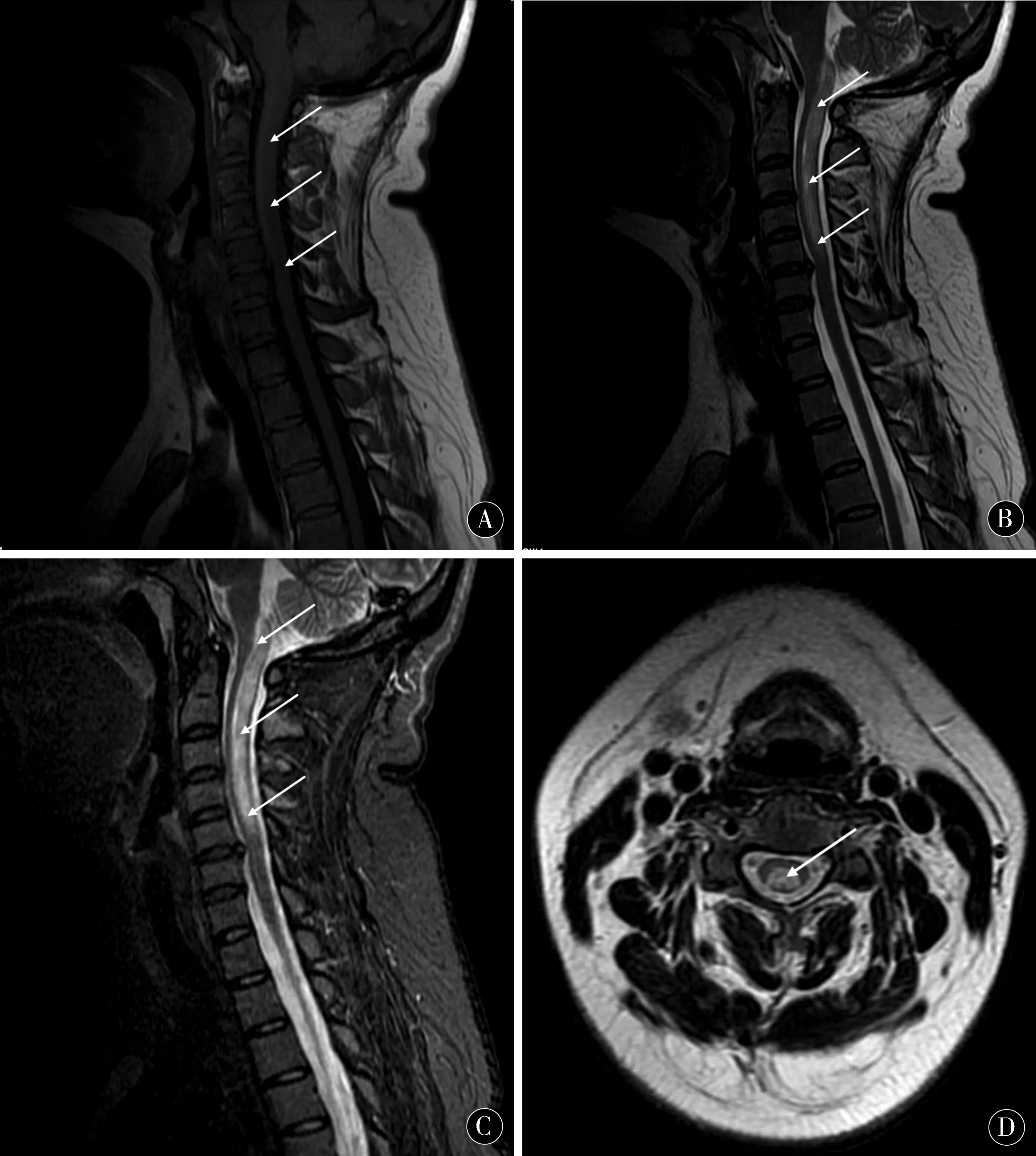
图1 颈椎MRI(2017-04-23)示延髓至C5水平脊髓长T1、长T2异常信号影(白色箭头指示)Figure 1 Cervical MRI (2017-04-23) shows abnormal signal shadows of long T1 and long T2 in the medulla oblongata to C5 (indicated by white arrow)
APQ4抗体被认为是NMOSD的血清生物学标志物,APQ4抗体导致的星形胶质细胞损伤是NMOSD的主要发病机制[14]。文献[15]报道,约90%的NMO及一半以上的NMOSD患者AQP4抗体阳性。APQ4是中枢神经系统中最丰富的水通道蛋白之一,丰富表达于毗邻微血管和软脑膜的星形细胞足突以及缺乏血脑屏障的下丘脑以及脑室周围结构[16]。临床影像学证实,NMOSD脑部病灶通常位于延髓背侧/最后区、脑干室管膜周围、间脑以及大脑侧脑室周围白质[17-18],说明APQ4高表达的区域是NMOSD典型病灶的分布区。但也有文献报道,10%~40%的NMOSD患者即便在发作期、治疗前、应用当前最敏感的检测方法仍无法检测出APQ4抗体[19]。目前,国内外诸多学者针对APQ4抗体阳性与APQ4抗体阴性的NMOSD患者进行比较得出结论:(1)抗体阳性的患者中女性发病比例高于男性[20-22]。(2)APQ4抗体阴性的NMOSD患者神经功能缺失症状较轻[23]。(3)APQ4抗体阳性的NMOSD患者累及脊髓范围可能更广泛[24-26]。(4)APQ4抗体阳性的NMOSD患者复发风险高[27-28]。该患者为APQ4抗体阴性,病变累及的脊髓范围较大,但神经功能缺失症状轻,恢复较好,随诊1 a无复发。除该患者累及脊髓范围较广外,其余与上述结论一致。针对该患者APQ4抗体检测结果阴性,主要考虑以下原因:(1)样本存留时间长:样本留存时间对于AQP4抗体的检测至关重要,而我院检验科尚未开展APQ4抗体检测,需外送检查,不排除标本存留时间及保存不当等因素。(2)免疫抑制剂的应用:患者在进行抽血及脑脊液检查时已行丙种球蛋白治疗,免疫抑制剂可抑制体液免疫及细胞免疫,中和致病性自身抗体、细胞因子及补体[29],故可导致检测结果阴性。(3)检测方法不同可能导致结果差异:目前,检测APQ4抗体常用检测方法有酶联免疫吸附试验(ELISA)、基于细胞测定法(CBA)、间接免疫荧光法(IFA)等,推荐检测方法为CBA法,该患者为CBA法,检测结果阴性。MAJED等[30]研究显示,荧光活化细胞分类法(FACS)检测脑脊液AQP4抗体的敏感性高于CBA法。尚有待进一步证据支持。(4)可能存在其他复发机制[31-34],有待进一步研究。
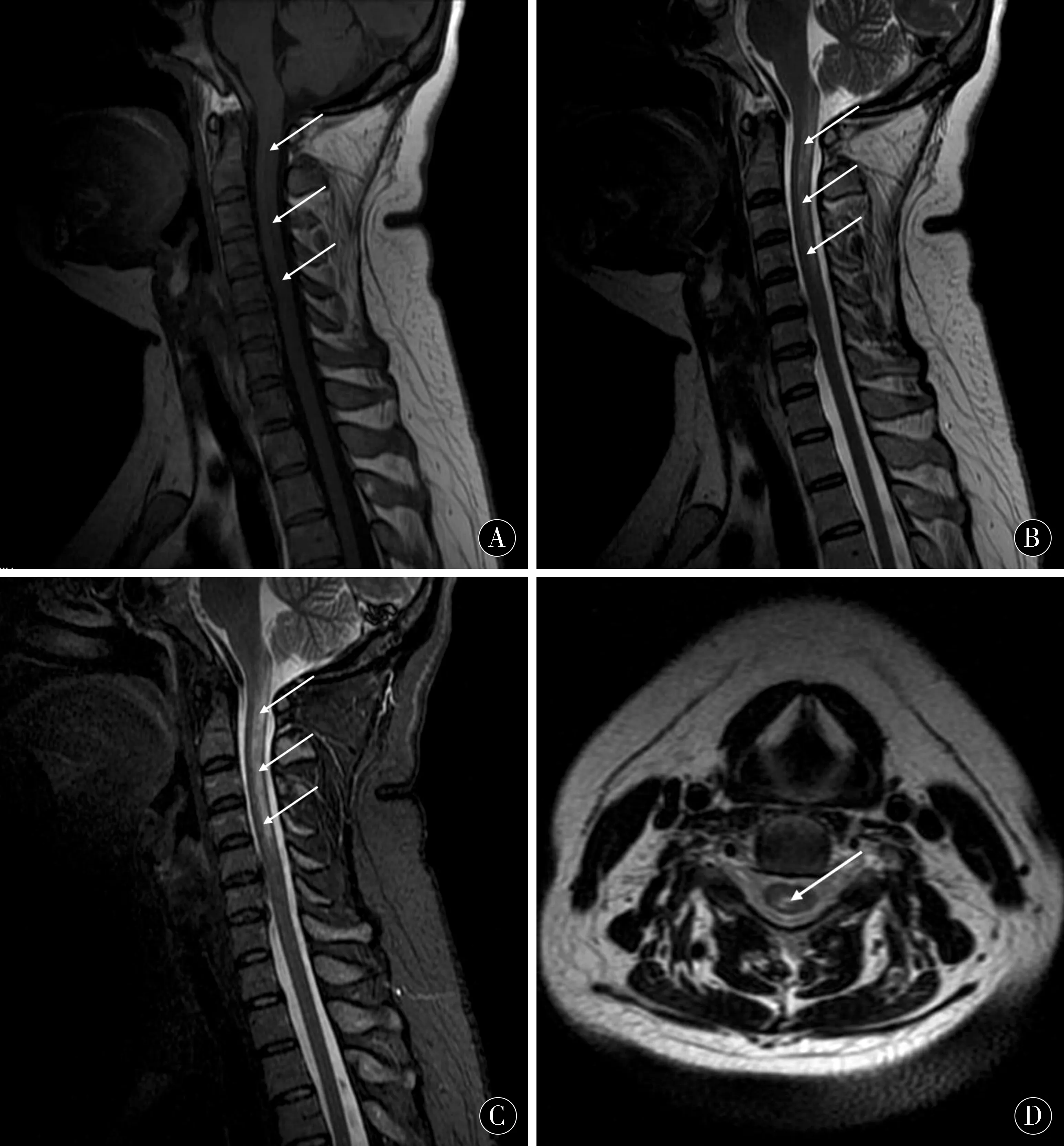
图2 颈椎MRI(2017-05-12)示延髓至C5水平脊髓长T1、长T2异常信号影(白色箭头指示)较前病灶明显缩小Figure 2 MRI of the cervical spine (2017-05-12) showed that the abnormal signal shadows of long T1 and long T2 in the medulla oblongata to C5 were significantly smaller than those of the anterior lesions
近几年来,针对NMOSD髓鞘少突胶质细胞糖蛋白(MOG)抗体成为很多学者的研究热点,认为APQ4抗体阴性的NMOSD患者血清中存在较高的MOG抗体表达[35-37],MOG抗体可能为其致病抗体。MOG抗体属于免疫球蛋白超家族,是中枢神经系统少突价值蛋白和髓鞘表面特异表达的蛋白[38]。研究证实,人类MOG抗体可以通过激活细胞抗体和补体介导的细胞毒性作用,加重髓鞘脱失[39]。SATO等[15]指出MOG抗体阳性的NMOSD患者具有以下临床特点:(1)好发于男性,男女比例约为1∶0.6,儿童多见。(2)实验室检查:多合并脑脊液寡克隆区带阳性。(3)CNS病灶分布:多累及双侧视神经,视盘水肿显著;脊髓病灶多累及下胸段或腰段,可见脊髓圆锥受累,脊髓水肿较APQ4抗体阳性患者显著,颅内病灶多累及深层灰质核团以及第四脑室周围。(4)治疗:激素治疗反应好,少部分患者具有激素依赖性,停止激素后较易复发。(5)预后:临床发作次数较少,复发率低,发作后神经系统功能缺损症状恢复更好。从上述临床特点看患者青年女性,非MOG抗体阳性好发人群,脊髓病灶以延髓,颈髓为主,非MOG抗体阳性脊髓好发部位,与SATO等的研究一致。
本例患者为自然流产后10余天开始出现后枕部麻木、疼痛,逐渐进展的双上肢麻木、无力症状,考虑与妊娠相关发作。SHIMIZU等[40]将“与妊娠相关发作”定义为妊娠期或分娩1 a内的发作,并发现约46.8%的日本NMOSD女性患者出现与妊娠相关的临床发作,包括首次发作或复发。NOUR等[41]研究发现,在NMOSD发病前后3 a期间发生流产的风险均较高。NMOSD发病后妊娠是流产的独立危险因素,而妊娠前疾病越活跃,发生流产的风险就越高。
NMOSD与妊娠流产关系密切,两者相互影响。目前多数学者认为AQP4抗体可能与妊娠相关复发及流产相关。妊娠期间中枢神经系统AQP4抗原表达上调[42],妊娠期间Th2为主的免疫改变也有利于APQ4抗体产生[43]。本例患者为流产后20 d左右出现NMOSD复发。目前认为抗NMO抗体、抗MOG抗体阳性的NMOSD复发风险高,抗NMO抗体阳性的女性患者妊娠期发生流产的风险高,但本例患者完善血清及脑脊液抗NMO抗体、抗MOG抗体均阴性,提示本病复发除与APQ4抗体有关外,尚有其他可能的发病机制。如妊娠期间母体的性激素水平在维持妊娠同时,对免疫系统产生作用,促使Th0淋巴细胞发育成Th2细胞,体液免疫功能增强,而Th1细胞及其细胞因子分泌被抑制,细胞免疫功能减弱[44]。妊娠可能通过性激素水平变化,增强了Th2细胞免疫及体液免疫而诱发NMOSD复发。但具体妊娠期雌激素水平、孕激素水平,免疫相关细胞因子,如TH1、TH2变化是否和NMOSD复发相关尚需要大样本研究。
妊娠流产是导致NMOSD复发的一个相关因素,但具体的发病机制尚不明确,NMOSD与妊娠流产关系密切,两者相互影响。本例血清及脑脊液APQ4抗体、MOG抗体均阴性,说明本病复发除与APQ4抗体、MOG抗体有关外,尚有其他可能的发病机制,需要临床更深入的研究。
猜你喜欢
军事文摘(2022年8期)2022-11-03
现代仪器与医疗(2022年1期)2022-04-19
昆明医科大学学报(2021年12期)2021-12-30
昆明医科大学学报(2021年4期)2021-07-23
云南医药(2021年3期)2021-07-21
中华养生保健(2020年5期)2020-11-16
保健文汇(2020年2期)2020-08-24
科教新报(2019年26期)2019-09-10
电子制作(2019年7期)2019-04-25
现代畜牧科技(2016年5期)2016-10-21
