
利用二代测序数据探索SPRY基因家族与中国人群非综合征型唇腭裂的关联
2019-06-18 10:25:36郑鸿尘李文咏王梦莹王斯悦周治波朱洪平
北京大学学报(医学版) 2019年3期
周 仁,郑鸿尘,李文咏,王梦莹,王斯悦,李 楠,李 静,周治波,吴 涛,朱洪平△
(1.北京大学公共卫生学院流行病与卫生统计学系,北京 100191; 2.北京大学口腔医学院·口腔医院,口腔颌面外科 国家口腔疾病临床医学研究中心 口腔数字化医疗技术和材料国家工程实验室 口腔数字医学北京市重点实验室,北京 100081;3.北京大学口腔医学院·口腔医院儿童口腔科,北京 100081)
非综合征型唇腭裂(non-syndromic oral clefts, NSOCs)是我国常见的出生缺陷之一,其患病率仅次于美洲印第安人和日本人[1],患病率每1 000名活产儿约为1.39~1.46[2]。NSOC根据累及解剖部位不同可分为3种亚型,即单纯腭裂(non-syndromic cleft palate only, NSCPO)、单纯唇裂(non-syndromic cleft lip only, NSCLO)和唇裂合并腭裂(non-syndromic cleft lip and palate, NSCLP);后两者由于流行病学特征及胚胎发育起源特征的相似性被认为具有相同的病因背景,在该领域病因学研究中通常合并为一类,称为非综合征型唇裂合并或不合并腭裂(cleft lip with or without cleft palate, NSCL/P)。NSCL/P是一种复杂性疾病,其遗传病因探索一直是该领域的研究热点。近年来广泛开展的全基因组关联研究(genome-wide association study,GWAS)已发现数十个影响NSOC发病风险的基因或区域[3-6],然而这些基因或区域仅能解释NSOC遗传度的约20%[7],提示仍有遗传危险因素尚未被发现。
由于GWAS的理论基础是常见复杂疾病的遗传危险因素来源于人群中弱势等位基因频率(minor allele frequency, MAF)大于等于5%的常见遗传变异[8-9],该方法难以探索MAF小于5%的罕见遗传变异或低频遗传变异,而MAF小于5%的罕见遗传变异或低频遗传变异可能具有更强的致病效应,对遗传度的贡献更大[10]。因此,搜寻致病性罕见变异是后GWAS时代非综合征型唇腭裂遗传病因学研究的焦点。二代测序技术(next generation sequencing,NGS)则为搜寻罕见致病位点提供了高效可靠的方法。该技术不仅能检测常见变异,更重要的是可以弥补GWAS难以覆盖到的罕见变异。根据测序覆盖范围不同,二代测序可以分为全基因组测序、全外显子组测序(whole exome sequencing,WES)和目标区域测序,其中WES覆盖整个外显子组,发现编码区的遗传变异更有利于对结果的生物学功能进行合理的解释,因此,全外显子组测序在复杂疾病的遗传病因探索方面具有一定的优势。此外,亲源效应也是尚未被解释的遗传度的可能来源之一,由于亲源效应是指亲本来源染色体上的等位基因出现差异性表达,即相同等位基因对疾病风险的影响因其亲本来源不同而不同,探索亲源效应不仅能加深对该疾病遗传病因的理解,还能为今后的唇腭裂遗传咨询、风险评估工作提供科学依据。
SPRY基因家族包含4个基因,分别为SPRY1、SPRY2、SPRY3、SPRY4,且位于不同的染色体。SPRY基因编码的蛋白质能够参与和调节多个涉及人体生长发育信号通路的正常表达,如RTK(receptor tyrosine kinase)信号通路和FGF(fibroblast growth factors)信号通路[11-12],其中,FGF信号通路能够调控细胞的增殖、分化和胚胎的形态学特征,曾被报道与唇腭裂的发生有紧密联系[13]。Ludwig等[14]在一篇Meta分析首次报道了SPRY2附近区域rs8001641的变异与NSCLP关联;随后Jia等[15]和Moreno Uribe等[16]在欧洲人群中成功复制了rs8001641位点的变异与NSCLP的关联。在中国人群中,Yu等[6]利用汉族人群3 379名NSCLP病例和8 593名对照者发现了SPRY2基因(rs9545308)与NSCLP的关联,并报道了该家族另一基因SPRY1基因的rs908822位点变异与NSCLP的关联,提示SPRY基因家族可能在中国人群NSCL/P发病过程中发挥作用。然而,目前在中国人群NSCL/P中对SPRY基因家族的分析多利用GWAS数据,对该基因家族中的罕见变异探索较少见。
本研究基于病例-双亲设计,以2016—2018年于北京大学口腔医院募集的 183个中国人群非综合征型唇裂合并或不合并腭裂核心家系(549人)为研究对象。利用其二代测序数据中的SPRY基因家族相关信息,展开单核苷酸多态性(尤其是罕见变异)和亲源效应分析,探索SPRY基因家族中的单核苷酸多态性及亲源效应与中国人群非综合征型唇腭裂发病风险的关联。
1 资料与方法
1.1 研究对象
本研究开始前获得北京大学医学伦理委员会审查批准(IRB00001052-15081),所有研究对象均签署知情同意书,未成年人由其监护人签署知情同意书。
本研究2016—2018年于北京大学口腔医院募集的183个中国NSCL/P核心家系(共549人)为研究对象。
纳入标准:(1)非综合征型唇腭裂患儿;(2)患儿及其父母自愿参加本次研究并且签署书面知情同意书。
排除标准:患儿父母中仅一方可以参加本次研究。
由临床专家和遗传学专家共同确诊病例及疾病亚型,所有病例通过临床检查排除综合征型唇腭裂。所募集的183名NSCL/P患者中包含114名男性,69名女性。本研究采用二阶段设计,于第一阶段对24个具有阳性家族史的NSCL/P核心家系进行全外显子测序阶段,第二阶段在159个NSCL/P独立样本中对第一阶段阳性信号进行验证。两阶段中NSCL/P患者的性别分布见表1。
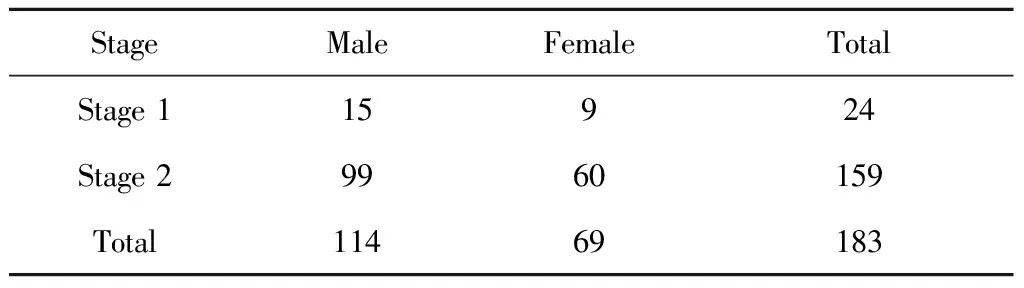
表1 中国人群183个NSCL/P患者的性别分布情况Table 1 The sex distribution of 183 NSCL/P cases in Chinese populations
1.2 数据收集及基因型检测
募集现场由经过统一培训的调查员通过面对面的问卷调查收集患者及父母的基本信息、环境暴露等情况,并根据临床体格检查获取患者的生长发育情况。对患儿及其父母采集每人4 mL静脉血,采用盐析法从血细胞中提取DNA样本进行基因型检测。第一阶段的全外显子组测序由武汉华大医学检验所有限公司完成。使用NimbleGen SeqCap EZ V3(64M)平台进行外显子组捕获,使用Illumina Hiseq4000测序平台进行高通量测序。根据测序深度(剔除测序深度小于6或大于500的位点)、基于样本的缺失率等指标对测序数据进行质量控制,得到一阶段分析的遗传变异位点。第二阶段对验证位点的检测采用KASP(Kompetitive Allele Specific PCR)技术,整个实验步骤按照LGC公司的KASP操作指南(www.lgcgenomics.com)进行。对二阶段验证位点的质量控制包括:(1)剔除分型缺失率>10%的SNP位点;(2)在父母亲人群中计算每一个SNP位点的MAF,剔除MAF<0.1%的SNP位点。
1.3 统计学分析
本研究所基于的二代测序研究采用二阶段设计,因此数据分析分为相应的两部分:第一阶段对SPRY基因的外显子组测序结果进行遗传变异的注释、功能预测及统计学分析,统计学分析包括单位点分析和多位点分析,结合以上结果挑选可能的致病位点作为二阶段验证位点;第二阶段对一阶段发现的位点进行单位点分析和亲源效应分析。
1.3.1一阶段数据分析
1.3.1.1遗传变异的注释及功能预测 本研究采用SnpEff(http: //snpeff.sourceforge.net/SnpEff_manual.html)软件对测序结果做注释及功能预测,包括变异所在区域,是否导致蛋白编码和氨基酸改变,Polyphen2、SIFT、CADD(combined annotation dependent depletion)分值等功能预测指标。Polyphen2用于评价特定位点变异引起的氨基酸变化对人体蛋白质结构及功能的影响,其取值范围为0~1,Polyphen2的取值越大表示该位点变异带来的氨基酸变化对人体蛋白质结构及功能的影响越大。根据Polyphen2取值大小可将变异分为良性(0~0.446)、可能有危害(0.447~0.908)、很可能有危害(0.909~1)3类。SIFT用于评价某个氨基酸变化对蛋白质功能的影响,通过在基因组中寻找与目标位点附近序列相似的序列,与其进行比对并计算发生碱基替换的概率,若位点发生碱基替换的概率小于0.05,则认为该位点是有害的。CADD分值也叫做C-scores,该指标将多个注释及功能预测指标整合为一个指标,可定量评价某位点变异带来的功能学影响以及是疾病致病位点的可能性大小,若某位点的分值大于或等于10,则认为该位点可能为疾病的致病位点。
1.3.1.2单位点分析 采用TDT方法进行单位点关联分析,其无效假设是杂合子父母传递任意一等位基因给患病子女的概率为50%,若研究人群中传递情况与无效假设不符,表明该等位基因与疾病存在关联,提示该遗传标记位点与潜在致病位点之间存在连锁或二者处于连锁不平衡。由PLINK (v1.07, http: //pngu.mgh.harvard.edu/purcell/plink/)软件完成。
1.3.1.3多位点分析 采用以家系为基础的序列核心关联检验(family-based sequence kernel association test)方法,将单个基因内多个遗传位点的效应进行整合, 以基因或区域为单位提供效应值,从而检验位点与表型之间的关联,此分析由R软件的famSKAT-RC包完成,该计算包在整合多个遗传位点的效应值时可同时纳入常见变异及罕见变异。
根据以上分析中遗传变异的注释结果、功能预测及统计学分析结果,选取满足以下任意一种情况的位点进入验证阶段:(1) Polyphen2≥0.909或SIFT≤0.05或CADD≥10的位点;(2) TDT中P值小于0.05的位点;(3)famSKAT中P值小于0.05的基因上的位点。
1.3.2二阶段数据分析
对纳入验证阶段的位点进行如下分析;本阶段分析采用Bonferroni法进行多重检验矫正,矫正后显著性水平P=0.05/3=0.017。
1.3.2.1单位点分析 采用TDT方法进行单位点关联分析,由PLINK (v1.07, http: //pngu.mgh.harvard.edu/purcell/plink/)软件完成。
1.3.2.2亲源效应分析 本研究采用Z检验分析亲源效应。原理为分别考虑父亲和母亲向子代传递中的传递不平衡情况,得到在父亲-子代、母亲-子代两种传递方向中某等位基因传递与未传递的比值比,通过比较母亲和父亲分别向子代传递过程中的比值比是否存在差异,以检验是否存在亲源效应。统计分析由PLINK(v1.07, http: //pngu.mgh.harvard.edu/purcell/plink/)软件完成。
2 结果
2.1 一阶段分析结果
本研究按照所制定的测序数据质量控制标准,对一阶段24个NSCL/P核心家系全外显子组测序数据中位于SPRY基因家族中的24个位点进行质量控制,共22个位点纳入分析。
位点注释结果显示本研究纳入的22个位点中,11个位于3′端非翻译区、1个位于5′端非翻译区、5个位于内含子区域、3个位于基因下游区、2个为错义突变位点。错义突变的位点有位于SPRY1基因rs1298215244位点(c.920T>C),其SIFT值为0.013,Polyphen2值为0.844,CADD值为4.46;位于SPRY2基因的rs504122位点(c.316C>T突变), 其SIFT值为0.097,Polyphen2值为0.001,CADD值为0.16。利用TDT在22个位点中进行单位点分析,结果显示22个位点均达不到统计学显著性水平(P<0.05), 其中P值最小的位点为SPRY1基因上的rs300574(P=0.083),P值最小的前五位位点的TDT检验结果见表2。
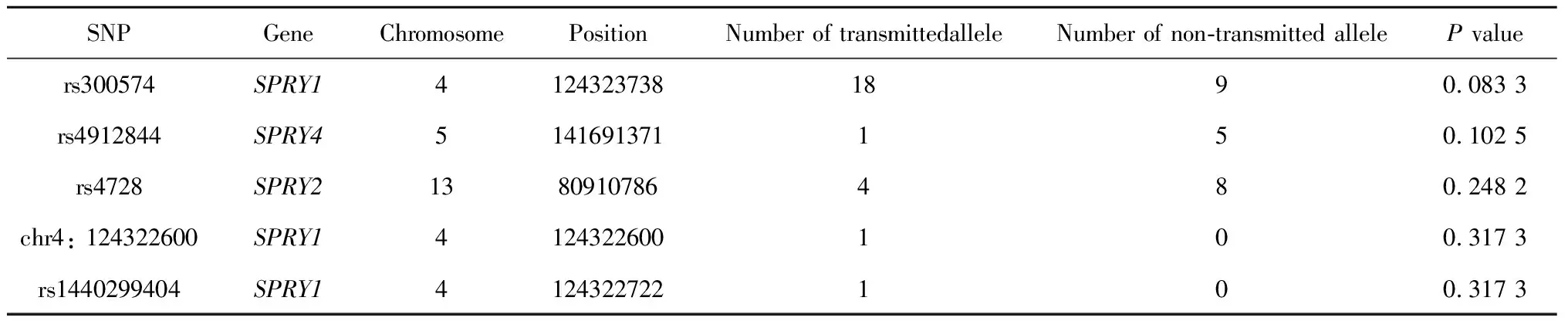
表2 中国人群24个NSCL/P核心家系SPRY基因家族中单位点的传递不平衡检验结果Table 2 Transmission disequilibrium tests of SNPs in the SPRY gene family among 24 Chinese NSCL/P trios
采用以家系为基础的序列核心关联检验以基因为单位进行多位点统计学分析。纳入分析的位点中有5个位于SPRY1基因,5个位于SPRY2基因,12个位于SPRY4基因。将这些位点以基因为单位进行组合,分别检验SPRY1、SPRY2、SPRY4基因与NSCL/P发病风险的关联。当phi分别取0、0.2、0.5、0.8、1时,SPRY基因与NSCL/P的关联均不能达到统计学显著性水平(P<0.05),其中参数phi为罕见变异对疾病遗传度的贡献程度,当phi取1时表明该疾病遗传度全部来源于罕见变异,当phi取0时表明该疾病遗传度全部来源于常见变异。以基因为单位的多位点分析结果见表3。
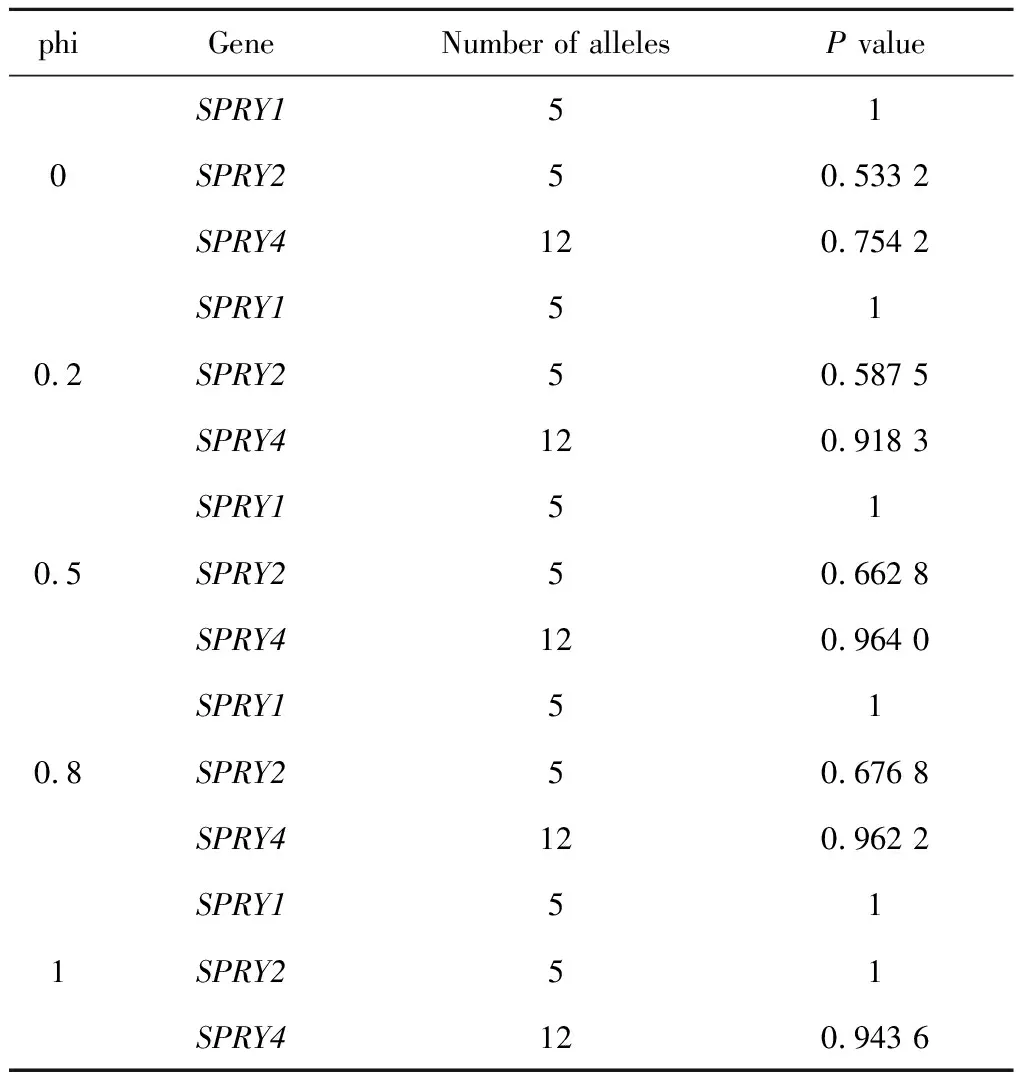
表3 中国人群24个NSCL/P核心家系中SPRY基因的以家系为基础的序列核心关联检验分析结果Table 3 The famSKAT analysis of SPRY genes among 24 Chinese NSCL/P trios
2.2 二阶段验证结果
根据一阶段测序数据分析结果及验证位点的纳入标准,并另外纳入一个错义突变位点,纳入二阶段验证的位点有两个,分别为rs1298215244(SPRY1)和rs504122(SPRY2)。对二阶段159个NSCL/P核心家系中这两个位点的检测结果分别进行单位点分析、亲源效应分析。
在159个NSCL/P验证家系中进行TDT检验,并利用Bonferroni法对结果进行多重检验矫正。由于纳入验证的rs504122(SPRY2)位点在中国人群中存在不同碱基的变异,分别为G>C和G>T,因此TDT检验次数为3次,Bonferroni多重检验校正后的统计学显著性水平为P=0.05/3=0.017,结果显示验证位点与NSCL/P发病风险的关联存在统计学意义,且达到经Bonferroni多重检验校正后的统计学显著性水平(P=0.05/3=0.017,表4)。
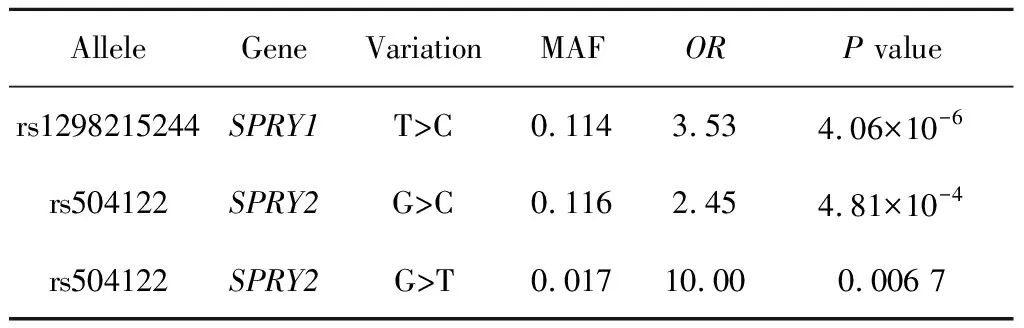
表4 中国人群159个NSCL/P核心家系中验证位点的传递不平衡检验结果Table 4 Transmission disequilibrium tests of potential signals in the SPRY gene family among 159 Chinese NSCL/P trios
MAF, minor allele frequency;OR, odds ratio.
在二阶段159个NSCL/P验证家系中对验证位点进行亲源效应分析,以探索遗传变异对疾病的作用是否受其亲代来源的影响,同样采用Bonferroni法对结果进行多重检验校正。结果显示位于SPRY1基因的rs1298215244其亲源效应与NSCL/P的关联具有统计学意义(P<0.05),但经Bonferonni校正后不能达到显著性水平(P=0.05/3=0.017)。验证位点亲源效应分析的检验结果见表5。

表5 中国人群159个NSCL/P核心家系中验证位点的亲源效应分析结果Table 5 Analyses of parent-of-origin effects for potential signals in the SPRY gene family among 159 Chinese NSCL/P trios
3 讨论
本研究基于病例-双亲核心家系设计,探索SPRY基因家族中单核苷酸多态性及亲源效应与中国人群NSCL/P发病风险的关联,发现SPRY基因家族中rs1298215244的T>C变异、rs504122的G>C变异两种常见变异和位于rs504122的G>T罕见变异与NSCL/P存在关联,未发现该基因家族内SNP位点具有亲源效应。
本研究所发现的两个单核苷酸多态性位点均位于SPRY基因的外显子区域,且位点变异类型为错义突变,说明该遗传变异可改变氨基酸编码,进而可能影响SPRY蛋白质的结构和功能。其中rs504122位点的突变与人体内多种组织的启动子组蛋白标记、增强子组蛋白标记及DNA剪切酶相关,且该位点的变异可能影响与KAP1蛋白的结合[17],该蛋白质已被发现与胚胎干细胞的分化过程存在紧密联系[18]。由于SPRY蛋白质为酪氨酸激酶受体(receptor tyrosine kinase, RTK)信号通路的拮抗剂,在多个生物学过程中发挥重要的调控作用[19-21];SPRY基因编码的蛋白质也是纤维母细胞生长因子(fibroblast growth factor, FGF)信号通路的调节因子[11],因此本研究所发现的位点可能通过影响SPRY蛋白质对胚胎生长发育的生物学过程,进而影响胚胎的颌面部发育。此外,曾有动物试验发现在小鼠体内的SPRY1和SPRY2基因的表达异常可影响胚胎的颌面部发育过程,导致面部裂、腭裂、鼻额骨无法形成等颌面部缺陷[22-24]。上述动物学试验发现也为本研究在SPRY基因家族外显子区域所发现的遗传变异位点提供了一定程度的支持,表明本研究所发现的变异可能通过影响SPRY基因的调控功能影响胚胎颌面部发育。
既往数项人群研究曾对SPRY基因展开非综合征型唇腭裂的遗传病因探索,Lugwig等[14]、Jia等[15]和Moreno Uribe等[16]曾先后在欧洲人群中发现并复制了位于SPRY2基因的rs8001641与非综合征型唇腭裂的关联,SPRY1与SPRY2在中国人群中也曾被发现阳性信号[6]。在该基因家族中罕见遗传变异的探索方面,Jia等[15]利用病例对照研究在病例组中发现了13q31.1区域的4个罕见变异,虽然其与唇腭裂的关联没有达到统计学显著水平,但这些罕见变异在对照组人群中均不存在,提示SPRY2基因中的罕见变异可能是该疾病的病因来源之一。上述研究所发现的位点多位于基因的内含子或基因间区域,较难对阳性信号进行生物学功能解释。尽管本研究发现单核苷酸多态性位点与既往发现的位点不重合且不存在强连锁不平衡,但本研究的阳性信号位于外显子编码区,提示其可能通过影响蛋白质功能和结构参与发病。
本研究未发现与NSCL/P具有关联的单核苷酸多态性位点具有亲源效应,提示阳性位点对疾病的影响程度不受等位基因的来源(父亲或母亲)所影响。亲源效应是指依靠单亲传递某些遗传学形状,亲本来源染色体上的等位基因出现差异性表达,即两个亲本等位基因中一方表达,另一方沉默的现象[25]。亲源效应分析的本质是探索同一等位基因对疾病发生风险的效应强度是否受该基因来源(来源于父亲或母亲)的影响。由于核心家系可收集患儿及其父母双方的遗传信息,核心家系研究是在亲源效应分析方面相对于病例对照研究具有研究设计上的天然优势。本研究未能发现SPRY基因的亲源效应与疾病发生风险相关,可能与样本量有限或亲源效应强度较弱有关,因此有关SPRY基因是否能通过亲源效应影响NSCL/P的发病风险仍有待后续研究的进一步探索。
综上所述,本研究以183个中国人群NSCL/P核心家系的二代测序数据中SPRY基因家族相关信息展开分析,可以进一步探索既往GWAS无法覆盖的罕见变异与NSCL/P的关系。研究基于病例-双亲核心家系设计,可以较好地控制人群分层所带来的混杂,并可借助父母及子代的遗传信息对基因检测结果进行质量控制,保证数据质量,同时为亲源效应分析提供可能。本研究所基于的测序研究采用二阶段设计,可提高研究效率;然而由于一阶段样本量较少,可能影响本研究发现罕见变异的功效;第一阶段未采用严格的多重检验校正以避免遗漏潜在致病位点,因而第一阶段结果存在假阳性的可能,但本研究在第二阶段采用了更大样本量的独立样本对第一阶段结果进行验证,并对阳性阈值进行了多重检验校正,可进一步筛选阳性信号,减少了被成功验证的位点中出现假阳性的可能。此外,测序数据中位于SPRY基因区域的位点数目较少,说明测序数据在该目标区域内的覆盖率较低,可能遗漏一些与NSCL/P发生风险关联的位点。本研究对NSCL/P亚型进行遗传病因探索,由于不同亚型其病因来源存在一定差异,还可依据是否患有腭裂、是否合并缺牙等对该亚型进行更加细致的划分,有助于提高研究效率。未来可在扩大样本量的基础上,针对SPRY基因家族进行目标区域测序,提高该区域的位点覆盖率,并对疾病亚型进行划分,更加全面、细致地探索SPRY基因家族中罕见变异与中国人群NSCL/P的关联,为未来进一步开展风险预测、遗传咨询及产前筛检、制定有效的预防策略措施提供科学依据。
猜你喜欢
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
当代陕西(2019年15期)2019-09-02 01:52:00
学苑创造·A版(2018年11期)2018-02-01 06:29:20
读者(2017年5期)2017-02-15 18:04:18
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
百科知识(2015年18期)2015-09-10 07:22:44
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
