
人GINS2基因RNA干扰慢病毒载体的构建及其在上皮性卵巢癌中的表达
2019-06-17 07:37严婷叶艾竹江恩利王玉娟
贵州医药 2019年5期
严婷 叶艾竹 江恩利 王玉娟
(贵州省人民医院(1.妇科;(2.检验科,贵州 贵阳 550002)
上皮性卵巢癌(Epithelial Ovarian Cancer,EOC)作为女性生殖系统中致死率最高,预后差的一种恶性肿瘤[1],其发生发展机制尚未完全阐明。我们此前的研究发现,GINS2基因在上皮性卵巢癌组织及细胞株中高表达。GINS2基因又称Psf2,是定位于16号染色体长臂上2区4带,全长1196bp,编码相对分子质量为21 000的蛋白质[2]。研究发现,GINS2在真核细胞中可以通过各种受体、生长因子和蛋白参与细胞周期的调控,并对细胞DNA的复制起始及增殖起到十分关键的作用[3]。GINS2在乳腺癌[4]、白血病[5]、肺癌[6]等恶性肿瘤组织中表达明显升高,提示GINS2在肿瘤的形成、增殖、转移中起重要作用。RNA干扰(RNA interference,RNAi)是由核苷酸的小分子干扰RNA(small interference RNA,siRNA)诱导细胞同源基因mRNA降解、引发靶mRNA 降解而导致基因表达沉默的过程。RNAi作用机制具有普遍性、特异性、高效性、位置效应等特点,已被广泛应用于基因功能研究[7]。本研究拟构建稳定干扰人GINS2基因的慢病毒载体,并检测其感染效率,为进一步研究GINS2在上皮性卵巢癌发生过程中的作用奠定基础,旨在探讨其作为靶向治疗的可能性。
1 材料与方法
1.1 材料及试剂 GV115慢病毒重组载体(hU6-MCS-CMV-EGFP)、慢病毒包装载体pHelper 1.0、pHelper2.0(上海吉凯基因化学技术有限公司,GeneChem),293T细胞株(CBTCCCAS,Shanghai,China),SKOV3细胞株(CBTCCCAS,Shanghai,China),DMEM(Hyclone)、胎牛血清(上海微科生化试剂有限公司),吉凯转染试剂(上海吉凯基因化学技术有限公司,GeneChem)、限制性内切酶(NEB)、T4DNA连接酶(Fermentas)、Taq酶(Vazyme)、质粒DNA提取试剂盒(TIANGEN)、SYBR Real-time PCR 试剂盒(Takara,Dalian,China)、鼠抗Flag抗体(Sigma),鼠抗GAPDH抗体(Santa-Cruz Biotechnology),羊抗鼠抗体(Sc-2005,Santa-Cruz Biotechnology)。
1.2 实验方法
1.2.1 GINS2 RNAi慢病毒表达载体的构建 根据GenBank 注册的GINS2(NM-016095)序列,根据RNAi序列设计原则,以GINS2基因为模板,设计多个19-21ntRNA干扰靶点序列。经设计软件评估测定后,选取以下序列作为干扰靶点:GATTAACCTGAAACAAAGA,GC%为31.60%。并使用阴性对照序列,Scramble序列:TTCTCCGAACGTGTCACGT。依据茎环结构的特点,分别合成2对短发夹RNA(short hairpin RNA,shRNA)单链,两端加上AgeI和EcoRI限制性内切酶位点。除此之外,在正链3’端添加TTTTT终止信号,而反链5’端添加终止信号互补序列。设计好的序列送至上海捷瑞生物工程有限公司合成相应的DNA oligo单链,退火形成具有粘性末端的DNA双链。然后运用 Fermentas T4连接酶与经过限制性内切酶AgeI和EcoR I双酶切后线性化的Hu6-MCS-CMV-EGFP载体连接。连接产物转化至TOP10大肠杆菌感受态细胞中,菌液均匀涂布于含有氨苄青霉素的LB琼脂平板上,37 ℃培养过夜。选取阳性克隆,PCR鉴定正确的质粒送测序。按照EndoFree Maxi Plasmid Kit说明书提取质粒,将质检合格的质粒进行病毒包装。
1.2.2 慢病毒颗粒的包装 转染前24 h,用胰蛋白酶消化对数生长期的293T细胞。培养24 h后,待细胞融合度达到70%~80%时,将重组慢病毒GV115载体20 μg, pHelper 1.0载体质粒15 μg及pHelper2.0载体质粒10 μg,与同体积的吉凯转染试剂混合均匀,调整总体积为1mL,室温下温育15 min。混合液缓慢加入293T细胞培养液中,于37 ℃、5% CO2细胞培养箱中培养。更换为完全培养液,培养48 h后,收集富含慢病毒颗粒的细胞上清液,离心、过滤取病毒浓缩液。分装后将病毒浓缩液-80 ℃保存于病毒管中。
1.2.3 慢病毒滴度测定 将293T贴壁细胞铺板,96孔板,每孔4×104个细胞的密度接种,慢病毒液按10倍浓度稀释5个浓度,将梯度稀释的含病毒培养基加入各孔中。4 d之后,观察荧光表达情况,经逐孔稀释及荧光共聚焦检测病毒滴度。病毒滴度=荧光细胞数/病毒原液量=2/(1E-6)=2E+6(TU/μL)=2E+9(TU/mL)。
1.2.4 稳定干扰GINS2表达的SKOV3细胞株的建立 选取培养生长状态良好的SKOV3细胞,接种于6孔培养板培养,24 h后每孔加入8 μL滴度为5×108TU/mL的shRNA慢病毒颗粒进行感染,采用含有10%胎牛血清的5A培养基。按照MOI=20,用慢病毒感染SKOV3细胞,感染后12 h,去掉上清液,换上新鲜的完全培养基。感染72 h后荧光显微镜下观察GFP表达情况,评估病毒对于SKOV3细胞的感染效率。
1.2.5 RT-qPCR检测GINS2基因mRNA的表达 收集细胞(6孔板80%细胞密度),采用Trizol法提取总RNA。用M-MLV Reverse Transcriptase(美国Promega公司)进行反转录获得cDNA。采用Real-time Quantitative PCR法检测GINS2基因mRNA的表达情况。PCR反应程序参照说明书进行。荧光实时定量PCR法检测GINS2的表达。选取GAPDH做内参,采用2-ΔΔCt法计算GINS2相对表达量。以GAPDH为内参目的基因。GINS2引物,Forward:5’-CAGAAATGTCGCCTGCTCC-3’;Reverse:5’-GGATTTCGTCTGCCTTCG-3’,扩增片段大小175bp。以GAPDH为内参基因,引物Forward:5’-TGACTTCAACAGCGACACCCA-3’;Reverse:5’-CACCCTGTTGCTGTAGCCAAA-3’,扩增片段大小121bp。
1.2.6 Western Blot 检测GINS2基因蛋白的表达 收集各组细胞,裂解液裂解细胞后提取总蛋白。取上清BCA法测定蛋白浓度。进行SDS-PAGE电泳,电泳结束后,将蛋白转移到PVDF膜上。以含5%脱脂牛奶的TBST溶液室温封闭PVDF膜1 h,加入一抗(鼠抗Flag,鼠抗GAPDH 1∶2 000)4 ℃过夜,TBST洗膜3次,每次10 min。加入羊抗鼠二抗(1∶2 000)室温下孵育PVDF膜2 h。充分洗涤后,ECL法显色,曝光成像,记录实验结果,判断GINS2蛋白的表达水平。
2 结 果
2.1 GINS2慢病毒干扰表达载体鉴定 体外合成的寡核苷酸上游单链和下游单链在体外退火后形成双链DNA,与线性化GV115载体连接,连接产物命名为psc24135。将连接产物转化大肠感受态细胞中。取150 μg菌液加入含有氨苄青霉素的LB培养基中过夜培养。挑取单个菌落为模板,进行PCR扩增,反应条件为:94 ℃ 3 min;94 ℃ 30 s;55 ℃ 30 s; 72 ℃ 30 s,22次循环; 72 ℃ 5 min。PCR结束后,取产物行电泳检测。psc24135电泳结果显示:泳道4、5、7、8是连接成功的shGINS2,而泳道6表示连接未成功 (图1)。由此判断泳道4、5、7、8为阳性克隆,保存泳道4的结果,命名为psc24135-1,并进行测序。DNA结果中显示shRNA片段已成功插入到Hu6-MCS-CMV-EGFP载体,说明成功构建针对GINS2基因的RNA干扰重组质粒,对阳性克隆进一步扩增菌液提取质粒用于病毒包装(图1)。
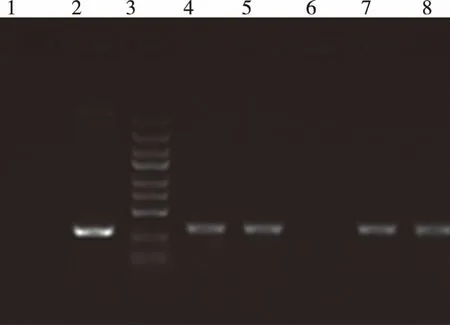
注:泳道1:阴性对照(ddH2O),泳道2:空载体自连对照,泳道3: Marker,泳道4~8:单克隆psc24135-1、2、3、4、5。电泳结果显示:泳道4、5、7、8是连接成功的shGINS2,而泳道6表示连接未成功。
图1 干扰载体的阳性克隆鉴定
构建的重组表达载体质粒经DNA直接测序法鉴定,与BLAST软件比对显示,插入的片段与GINS2 RNA干扰慢病毒载体质粒(psc24135-1)的干扰片段完全相符,见图2。
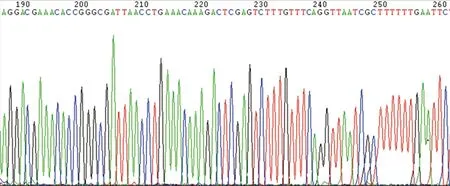
注:插入片段序列位于198~254。
图2 GINS2 RNA干扰慢病毒载体质粒的DNA测序结果
2.2 慢病毒载体感染293T细胞株以及慢病毒的包装和滴度测定 当293T细胞的生长状态达到产生慢病毒实验的要求时,将GINS2干扰质粒、慢病毒包装辅助质粒共转染293T细胞,48h后收集病毒粗提液进行浓缩,利用荧光法测定此靶点的病毒滴度,结果为5×108TU/mL,显示病毒成功包装,可用于后续研究。
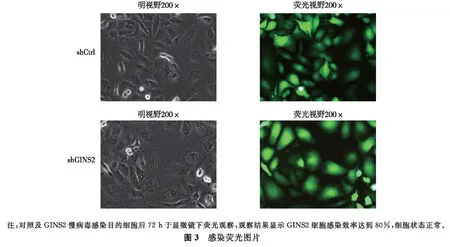
2.3 稳定干扰GINS2基因表达的SKOV3细胞的建立 含有GINS2基因RNA干扰序列的慢病毒对上皮性卵巢癌细胞株SKOV3细胞成功,感染后72 h左右,荧光显微镜观察报告GINS2基因的表达情况,荧光率即为阳性感染率;结果显示SKOV3细胞感染效率达到80%,细胞状态正常(图3)。
2.4 重组慢病毒对SKOV3细胞GINS2基因表达及蛋白表达的影响 RT-qPCR的方法和Western Blot法检测SKOV3细胞中GINS2基因敲减后mRNA的表达量及蛋白表达量,进而判断靶点的干扰效果。定量PCR结果可以看出,经shRNA慢病毒感染后,与对照组对比,实验组SKOV3细胞中GINS2基因在mRNA水平的表达量受到抑制(P=0.000 001,P<0.05),沉默效率达到91.7%(图4A)。从Western Blot的结果可以看出,GINS2组未检测到蛋白,靶点对GINS2基因的外源表达有明显沉默作用,因而是有效靶点(图4B)。
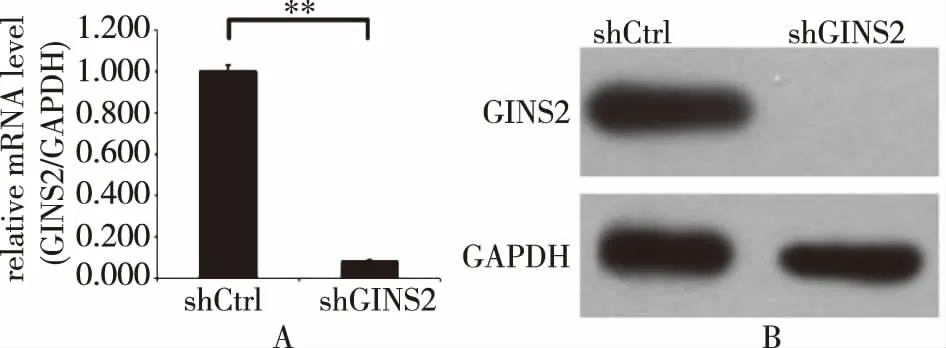
注:shCtrl为阴性对照组,shGINS2为shRNA组。4A:RT-PCR检测mRNA表达丰度,shGINS2组与shCtrl组相比**P<0.05。4B:Western Blot检测蛋白表达量的变化。
图4 GINS2基因敲减后mRNA及蛋白表达水平的比较
3 讨 论
GINS复合物(Go,Ichi,Nii,and San; five,one,two,and three in Japanese)是于2003年通过基因筛选的方法首次发现的一个核酸复制因子,它在体内形成一个环状结构,在核酸复制初期结合到染色体上并在核酸的延伸过程中发挥重要作用[8-10]。GINS复合物是一种复制解旋酶,在移动到复制叉前打开DNA双链。研究发现,在小鼠和酵母模型中,GINS复合体与微型染色体的修复相关,调节DNA复制的发生和发展。相继发现GINS复合体与人类DNA的复制相关[3]。也有文献表明,DNA复制相关蛋白在不同细胞中有不同的作用,如在决定中心体复制数量和疾病发生的不同阶段方面,GINS均发挥了一定功能,特别是与染色体的分离密切相关[11]。本研究前期结果显示,GINS2基因在上皮性卵巢癌组织和细胞株中均高表达,进一步明确GINS2基因在上皮性卵巢癌发生、发展中的作用及其分子机制将有助于发展上皮性卵巢癌的治疗措施,从而进一步提高患者生存率。
慢病毒为一类逆转录病毒的总称,由慢病毒改建而来的载体系统以高效且稳定的基因转移效率在基础和应用研究领域得到广泛应用。它能够实现持续的基因传递、将载体稳定的整合到宿主基因组;能够感染分裂和非分裂细胞;载体转导后并不表达病毒蛋白[12]。RNA干扰(RNA interference,RNAi)是指正常生物体内抑制特定基因表达的一种现象,在进化过程中高度保守的、由双链RNA诱发的、同源mRNA高效特异性降解的现象。其利用RNA介导,特异性地阻断和降低目的基因的表达[13]。将慢病毒载体作为RNAi技术的载体,可以结合两者优势,特异性抑制哺乳动物的各类细胞中基因的表达,成为基因功能研究和基因治疗的有力手段[7]。
本研究针对GINS2基因设计的特异性siRNA靶序列,成功构建了以GFP为报告基因的GINS2 shRNA慢病毒载体。采用GV115、pHelper 1.0及pHelper2.0三载体包装系统确保慢病毒载体系统安全性,该系统包括转移质粒、包装质粒和包膜质粒。病毒感染目的细胞不会再感染其他细胞,不会利用宿主细胞产生新的病毒颗粒。GV115载体中含有HIV的逆转录、包装、整合元件以及辅助元件,并带有GFP作为报告基因和puro抗性基因作为抗性标记,用于观察目的蛋白表达情况和筛选稳定感染的细胞株。pHelper 1.0载体中含编码病毒主要的结构蛋白、特异性的酶和相关调节因子的基因。pHelper2.0载体中含有提供病毒包装所需要的包膜蛋白的基因。病毒包装时,将这三种病毒包装质粒共转染入包装细胞293T,各载体产生病毒各组成蛋白,并包装成病毒颗粒。
PCR阳性克隆测序结果表明,GINS2基因成功插入GV115载体并重组为慢病毒载体GV115-GINS2-RNAi。将GV115-GINS2-RNAi质粒与病毒包装辅助质粒共转染293T细胞,结果显示,慢病毒可高效稳定的转入上皮性卵巢癌SKOV3细胞,感染效率达到80%。证明成功构建了GV115-GINS2-RNAi慢病毒载体,并获得GINS2慢病毒颗粒。病毒滴度可以反映病毒感染靶细胞的能力及效率,是评价包装细胞分泌感染性病毒颗粒的主要指标。
综上,本研究采用慢病毒介导RNAi技术构建特异靶向GINS2基因的慢病毒RNA干扰系统,为后续研究GINS2基因沉默后上皮性卵巢癌细胞生物学功能改变及分子机制的研究打下了坚实的基础。
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
数学大王·低年级(2020年8期)2020-08-14
生物工程学报(2019年1期)2019-01-30
中成药(2018年3期)2018-05-07
安徽医科大学学报(2016年12期)2017-01-15
小雪花·初中高分作文(2016年9期)2016-05-14
肿瘤预防与治疗(2015年2期)2015-09-26
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年4期)2015-06-10
中国当代医药(2015年16期)2015-03-01
