
Phylogeny of Culex theileri virus flavivirus in Spain, Myanmar, Portugal and Turkey
2019-06-11 02:04:44EleonoraCellaDomenicoBenvenutoDanieleDonatiFrancescoGarilliSilviaAngelettiStefanoPascarellaMassimoCiccozzi
Eleonora Cella, Domenico Benvenuto, Daniele Donati, Francesco Garilli, Silvia Angeletti,Stefano Pascarella, Massimo Ciccozzi
1Unit of Medical Statistics and Molecular Epidemiology, University Campus Bio-Medico of Rome, Rome, Italy
2School of Nursing, Faculty of Medicine, Department of Biomedicine and Prevention, Tor Vergata University, Via Montpellier 1, 00133, Rome, Italy
3Faculty of Medicine, University Campus Bio-Medico of Rome, Rome, Italy
4Unit of Clinical Laboratory Science, University Campus Bio-Medico of Rome, Italy
5Department of Biochemical Sciences "A. Rossi Fanelli", University of Rome “La Sapienza”, 00185 Roma, Italy
Keywords:Culex virus Flaviviridae Phylogenetic analysis Evolution
ABSTRACT Objective: To study the genetic diversity of Culex theileri flavivirus and the spread of this virus among Spain, Portugal and Turkey.Methods: A database consisting of 55 sequences of the NS5/3’UTR region of Culex theileri flavivirus group downloaded from GenBank were aligned and manual edited with Bioedit.ModelTest v. 3.7 was used to select the simplest evolutionary model that adequately fitted the sequence data. Maximum likelihood analysis was performed using MEGA7. The phylogenetic signal of the dataset was investigated by the likelihood mapping analysis.Results: The phylogenetic tree showed three clusters. Myanmar sequences clusterd together with Turkish sequences, Spain and Portugal strains grouped together and two Turkish sequences grouped separately. Selective pressure analysis showed a moderate percentage of sites (22.5%) under pervasive negative selection and only 1% under pervasive positive selection. The sites subject to selective pressure in CTFV RdRp NS5 fragments have been located onto the predicted three-dimensional structure.Conclusions: Phylogenetic and evolutionary analysis can be an important tool for understanding the evolutionary impact of the probable contemporary existence between nonpathogenic and pathogenic flaviviruses among these vectors.
1. Introduction
The genus flavivirus comprises more than 70 RNA virus species that include Yellow fever virus, Dengue virus, Japanese encephalitis virus, and the Tick-borne encephalitis virus complex. Many of these arthropod-borne viruses represent dangerous threats to human health and have been subjected to intensive research to unravel their molecular and virological properties[1].
Insect-specific flaviviruses (ISFs) are distinct for the inability to be propagated in vertebrate cell lines and lack of vertebrate hosts[2]. ISFs have been recognized as a major phylogenetic group along with mosquito-borne, tick-borne and no-known-vector flaviviruses[3-5]. ISFs share many similar properties with widely known pathogenic arthropod-borne flaviviruses such as dengue virus(DENV), West Nile virus (WNV) and tick-borne encephalitis virus(TBEV). They are enveloped, single-stranded RNA viruses, with a single open reading frame (ORF) that encodes the viral polyprotein,which is then cleaved by viral and host proteases to form three structural (C, preM and E), and seven nonstructural (NS1, NS2A,NS2B, NS3, NS4A, NS4B and NS5) proteins[6]. ISFs seem to have a global distribution and have been detected in field-collected mosquitoes from Europe, East Asia, Africa, the American Continent and Australia[2,7]. The ISFs possess potentially distinct and unique features among flaviviruses, which are likely to have an impact on the circulation of their related vector-borne pathogens. Their widespread distribution without obvious reservoirs or amplification hosts requires exploration of potential mechanisms, such as vertical transmission, contributing to their dispersion[7-9]. In particular, Culex(Cx.) theileri flavivirus (CTFV) seems to affect the flight activity of Culex mosquitos[10].
The aim of this study was to investigate the evolutionary patterns of Cx. theileri virus using all the available sequences from National Center for Biotechnology Information (NCBI). It has performed a phylogenetic analysis including phylogenetic tree and homology model analysis.
2. Materials and methods
2.1. Sequence dataset
A dataset were built based on previous investigations about Cx.theileri flavivirus species made on the NCBI website, the previous investigation found a clade consisting of 55 sequences of NS5/3’UTR region of Cx. theileri flavivirus which has been selected and downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank/). All the sequences were aligned and manually edited using the Bioedit program v7.2.5, as already described[11].
2.2. Phylogenetic analysis
The phylogenetic signal was investigated by means of the likelihood mapping analysis of 10 000 random quartets by using TreePuzzle program as already described[12]. In this analysis, groups of four randomly chosen sequences (quartets) were evaluated using Maximum Likelihood. For each quartet, the three possible unrooted trees were reconstructed under the selected substitution model. The likelihoods of each tree were then plotted on a triangular surface,so that fully resolved trees fall into the corners and the unresolved quartets in the center of the triangle (indicating a star-like signal).When using this strategy, if more than 30% of the dots fall into the center of the triangle, the data are considered unreliable for the purposes of phylogenetic inference[12].
The evolutionary model was chosen, as the best-fitting nucleotide substitution model in accordance with the results of the hierarchical likelihood ratio test implemented in MODELTEST software (version 3.7)[13]. The Maximum Likelihood tree was reconstructed using MEGA7 with Tamura-Nei model as evolutionary model selected by ModelTest[13-15].
Adaptive Evolution Server (http://www.datamonkey.org/) was used to find eventual sites of positive or negative selection using FEL (Fixed Effects Likelihood) which uses a maximum-likelihood(ML) approach to infer nonsynoymous (dN) and synonymous (dS)substitution rates on a per-site basis for a given coding alignment and corresponding phylogeny[16]. Sites were considered to have been subjected to statistically significant positive or negative selection based on P value < 0.05 (reference sequence used, accession number: KX652375.1).
Possible spreading ways from Spain, Portugal, Myanmar and Turkey have been analyzed using the website http://portugalturkey.com/ and http://www.mfa.gov.tr/turkey_myanmar-bilateraleconomicand-commercial-relations.en.mfa. for a better understanding of possible spreading paths of the vector of the virus.
2.3. Homology modelling
Sequence alignments and analyses were obtained through the Jalview editor[17] and the programs ClustalO[18] or Muscle[19,20].Structural templates homologous to the sequences studied within this work have been identified with HHpred[20] and SwissModel[21]template search tools. Homology models were built with the program Modeller[22] and validated with QMEAN[23] and ProsaⅡ[24] software tools. Three-dimensional structures were displayed and studied with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schroedinger, LLC) or Chimera[25]. Sequence conservation and mapping analysis has been carried out with the Consurf web tool[26].
3. Results
3.1. Phylogenetic analysis
The percentage of dots falling in the central area of the triangle was 4.6%, the dataset did not show more than 30% of noise and contained sufficient phylogenetic signal (Figure 1).
The phylogenetic tree showed three clusters (Figure 2). The cluster Ⅰ included 28 strains isolated from Myanmar and Turkey.Cluster Ⅱ included only two sequences from Turkey. The cluster Ⅲincluded 25 strains isolated from Spain and Portugal.
CladeⅠwas composed by two subclusters: 18 sequences all isolated in 2014 in Myanmar grouped in clusterⅠa and cluster Ⅰb included 10 sequences of Cx. theileri flavivirus isolated from 2014 to 2015 in Turkey.
ClusterⅡinclude only two Turkish sequences isolated in 2014 and 2015. Cluster Ⅲconsisted of two different clusters (Ⅲa and Ⅲb):Ⅲa included Spanish isolates (in 2006) with the sequences isolated in Portugal in 2012 and Ⅲb strains isolated in Spain and Portugal respectively from 2003 to 2006 and in 2012 (Figure 2).
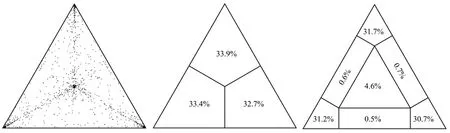
Figure 1. Likelihood mapping of Culex theileri flavivirus dataset. The dots inside the triangles represents the likelihood of the possible unrooted topologies for each quartet. Numbers indicate the percentage of dots in the centre of the triangle corresponding to phylogenetic noise (star-like trees).
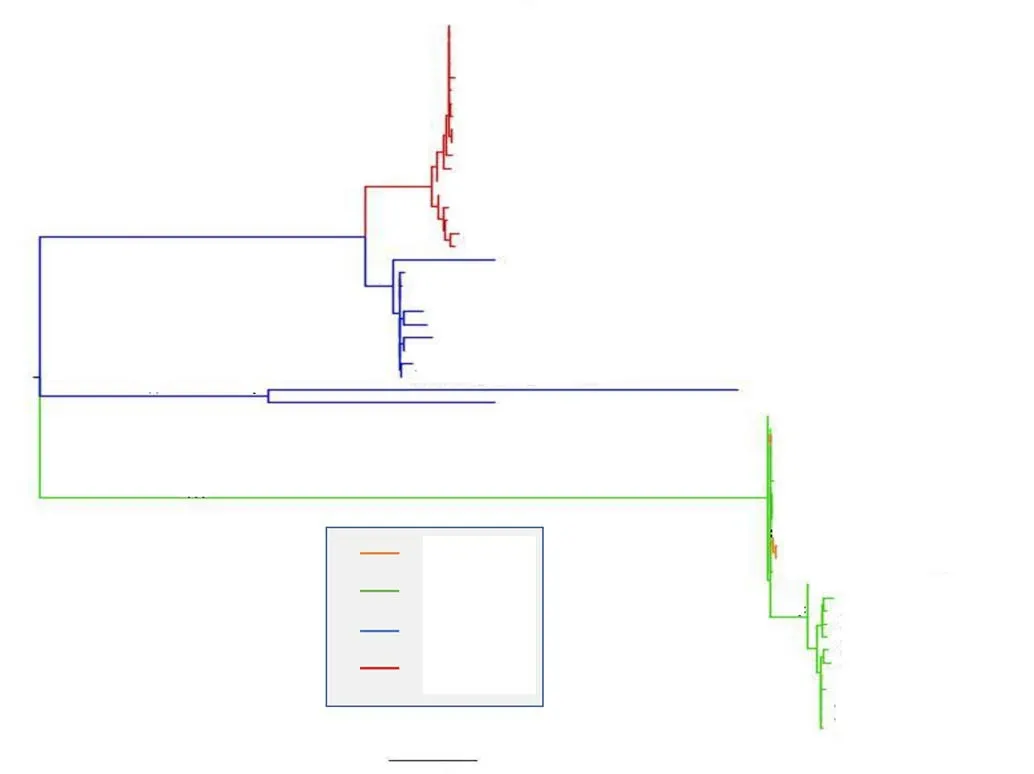
Figure 2. Maximum likelihood tree of Culex theileri flavivirus. Along the branches an asterisk (*) indicating a statistical value from bootstrap (>70%).
FEL analyzed 297 sites and found evidence of pervasive positive selection in 2 sites with P<0.05, it has been evidence of pervasive negative selection in 67 sites (22.5%) with P<0.05.
3.2. Homology modelling
A multiple sequence alignment among the RNA-dependent RNA polymerase (RdRp) fragments of CTFV NS5 proteins and the structurally solved NS5 sequences from flavivirus has been obtained(Figure 3). According to the alignment, the CTFV RdRp fragments here considered cover the region 488-798 of the full-length Japanese encephalitis virus NS5 (PDB code 4K6M). This portion (Figure 4)contains the “Palm”, “Middle” and part of the “Thumb” subdomains of the human right hand architecture characteristic of the NS5 proteins[27]. To map the structural variability of the variant CTFV RdRp fragments and their sites under selection pressure, homology modelling has been applied. Among the available structural templates, the PDB structure with code 4HDG corresponding to the crystal structure of the RNA-dependent RNA polymerase domain from Japanese encephalitis Virus NS5 in complex with GTP, has been selected. The decision has been taken in consideration of the suitable quality of the structure in terms of resolution (2.38 Å) and R-value (0.276), for the presence of the ligand GTP, and for the high sequence identity to the targets (about 53%). The sequence heterogeneity of the set of CTFV RdRp sample suggested to build models for at least two variants each representing a sequence subgroup (Figure 3). To this purpose, the fragments denoted by the codes HE997074 and KU958176 in Figure 3 have been selected.Models created from the two fragments do not show significant differences except for conformations of exposed loops (Figure 5);for that reason, the HE997074 model was used as reference.

Figure 3. Multiple sequence alignment between the fragments of Culex theileri flavirus RdRp domain of NS5 proteins and structurally solved homologous NS5 proteins from flaviviruses PDB codes of the RdRp domains are: 2HFZ (WNV); 4HDG (JEV); 4K6M (JEV-SA-14); 5JJS (DENV-3); 5WZ3 (ZIKV). Blue and green bars below the alignment indicate deletions and insertions in the CTFV RdRp sequences. Cyan arrows mark the CTFV RdRp sites under selective pressure. Labelled grey bars mark the conserved viral RdRp catalytic motifs. The red line shows the position of the “Middle finger” subdomain. Sequence position numbering refers to 4HDG structure.

Figure 4. Cartoon model of the chain A structure of the full-length Japanese encephalitis virus (PDB code 4K6M). The Methyltransferase (MTase)and RNA dependent RNA polymerase (RdRp) domains are labelled.Grey spheres indicate zinc ions, while sphere model in the MTase domain represents S-adenosyl-L-homocysteine. Dark red ribbon marks the CTFV RdRp fragments coverage of the full-length structure. Positions of the N- and C-terminal edges are labelled.
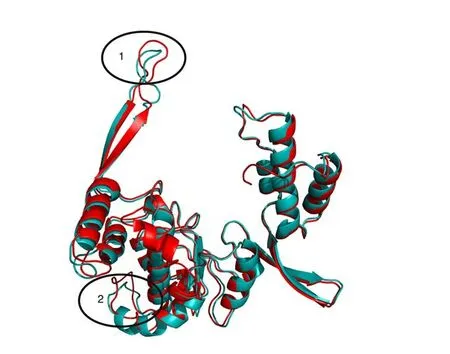
Figure 5. Structural superposition between the homology models of HE997074 (red cartoon) and KU958176 (cyan cartoon). Ovals enclose loops displaying conformational differences between the two models. Oval 1 corresponds to the “Middle” subdomain containing the position 567(Figure 3) under selective pressure and the insertion Asn-His (positions 584-585 in Figure 3). Oval 2 encloses the region of the large insertion in 4HDG(positions 636-644, Figure 3).
The sequence alignment in Figure 3 hints at the presence of several structural differences between the CTFV RdRp fragment set and the corresponding NS5 structures from flaviviruses. In particular, two deletions in the CTFV fragment set correspond to Gln residues in positions 528, and position intervals 636-644 of 4K6M and 4HDG sequences (Figure 6). In addition, two 2-residue insertions are between sequence positions 33-34 and 62-63 of the CTFV RdRp as reported in Figure 3, which correspond to positions 557-558 and 584-585 of 4K6M and 4HDG sequences (Figure 5).

Figure 6. Superposition between the homology model of CTFV RdRp NS5 with code HE997074 (red cartoon) and the structural template 4HDG (orange cartoon). Active site GTP is displayed as stick model. Grey and green spheres denote zinc and magnesium ions. Cyan and green sticks models indicate CTFV RdRp positions under selective pressure and insertions with respect to 4HDG. Orange sticks indicate the 4HDG insertion residues. Numbering refers to full-length 4HDG sequence. The two-residues insertion of CTFV RdRp between the 4HDG positions 557-558 and 584-585, respectively are indicated. Left figure is rotated by 180° along the vertical y axis with respect to that represented on the right panel.
The sites subject to selective pressure in CTFV RdRp NS5 fragments have been located onto the predicted three-dimensional structure (Figure 6). The two sites are at columns 44 and 134 of the alignment displayed in Figure 3 which correspond to positions 567and 655 of 4K6M and 4HDG structures.
Position 44 (567 of 4K6M) displays Ala or Ile residues in the flaviviruses sequences whereas it shows Ala in the structurally solved NS5 proteins. It is located within the -helix encompassed by positions 562-576 in 4K6M and it is buried with respect to the protein surface.
Position 134 in CTFV RdRp NS5 fragments (655 of 4K6M)displays Asn and, in two cases, His. The corresponding position in the structurally solved NS5 proteins has Asn or Lys. The position is within the “Palm” of the RdRp domain, exposed to the solvent, at a kink of a helix encompassed by 4K6M positions 642-662. In CTFV RdRp the N-terminal segment of the helix is deleted (Figure 3, 4)and replaced by a predicted loop.
ConSurf analysis conducted on the NS5 RdRp domain suggests that the position 44 is at a site conserved within homologous NS5 sequences from different flaviviruses (Figure 7). The conservation may reflect the stabilizing role of the hydrophobic interaction taking place between the Ala/Ile and other hydrophobic side chains from the surrounding protein environment (not shown). The position 134 sits at a site with generally variable residues within homologous NS5 sequences (Figure 7).
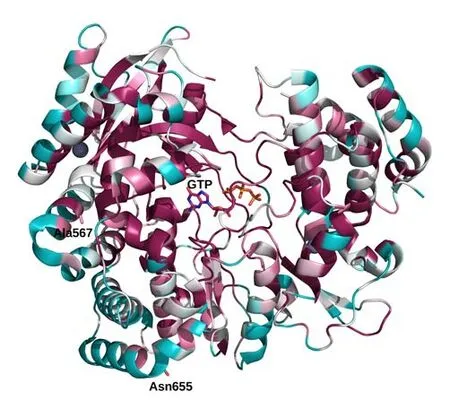
Figure 7. Cartoon depiction of the 4HDG structure coloured according to the conservation score calculated by ConSurf web tool. The colour scale ranges from cyan (high sequence variability) to dark red (high sequence conservation). White is the ”neutrality” colour. GTP is displayed with stick models. Positions subject to selective pressure are represented as stick models and labelled accordingly. All of them occur in highly variable positions of the RdRp domain of Flavivirus NS5 proteins.
4. Discussion
Flavivirus have a great impact on human health[28,29]. Many viruses assigned to the flavivirus genus are exclusively replicated and detected in insects and named ISF and in last years, an increasing number of newly discovered ISFs probably due to the bio surveillance efforts regarding arthropod-borne viruses and new sequencing techniques were observed[30].
Studies on phylogenetic relationships of ISFs in relationship with others accepted flaviviruses are not clear, and they have previously been suggested to form a sort of divergent outgroup suggesting the possibility to represent an ancestral lineage of flaviviruses
Despite statistically supported separation of the strains originating from Portugal and Turkey based on CTFV NS5 RdRp gene region,current information indicates that these strains constitute local variants of CTFV. Although the most comprehensive characterization of the CTFV was performed from specimens collected in Portugal during 2009 and 2010, there is evidence for the circulation of several related and potentially identical strains[7]. Partial NS5 sequences closely related to CTFV have been detected in Spain and Portugal from 2006 to 2010 with different acronyms (CxthFV) or names(Spanish Culex flavivirus, SCxFV)[31,32].
Conversely, some variations, especially found on the putative viral capsid, envelope and non-structural proteins NS1, NS2a, NS3, NS4b and NS5, are detected in strains from either location, which might be reflecting environmental adaptive processes.
Moreover, presence of partial CFTV NS5 sequences could also be detected in Culex pipiens pools in Turkey as well in Portugal and Spain on various occasions between 2006 and 2015[32-34].
Thus, the host range of CFTV may not be restricted to Culex theileri. Although a grouping of the ISFs according to their host mosquito species seemed plausible in initial analyses[35], several phylogenetically distinct ISFs have been repeatedly detected in various Culex and Aedes spp. Mosquitoes in different areas[7],suggesting independent introductions and multiple host-switching events[3]. As we can see from the data there is a consistent phylogenetic relationship among the presence of ISF in Spain,Portugal, Myanmar and Turkey, probably the infected Cx. theileri has spread by land from Spain to Portugal and from Turkey to Myanmar, vectored by mosquitos through commercial trading as so as happened with Aedes albopictus from Africa to Mediterranean areas in the recent years. Recently, passive surveillance activities to monitor mosquito species was performed in Europe since international trading based on tires is a well documented route of transmission of Aedes albopictus worldwide[36,37].
Selective pressure analysis showed a moderate percentage of sites(22.5%) under pervasive negative selection and only two sites (1%)under pervasive positive selection. The first position under positive selective pressure has two amino acidic variants (Ile and Ala),interestingly the Turkish and Myanmar strain have the same residue(Ile) and Spanish and Portuguese strains only Ala residue. Similarly,in the other position under selective pressure the Turkish strains have only His residue and Myanmar, Spanish and Portuguese strains only Asp residue. These results suggest a different distribution and different spread of CTFV strains between these countries.
Protein sequence comparison highlighted several sequence differences between the CTFV RdRp fragments and the reference structurally solved RdRp domains from flaviviruses. Although it is very difficult if not impossible to predict the functional and structural impact of these variations, it may be argued that since insertions and deletions occur mostly in exposed loops, they may alter chain flexibility and interaction with other molecular components. Of particular interest can be considered the insertion in the CTFV RdRp “Middle finger” loop (positions 584-585 of 4HDG), which contributes to the interaction of the domain with the connected MTase module. It has been suggested that the interplay is relevant for NS5 catalytic process[38]. One of the positions (site 567 of 4HDG)under selective pressure occurs in a buried conserved site within the helix encompassed by positions 562-576 of 4K6M. Conservation of the hydrophobic characteristic (only Ala or Ile occur in the CTFV sequences) strongly suggests that this site is important for providing stability through van der Waals contacts with other hydrophobic side chains from the adjacent helix. The second position under selection,(site 655 of 4HDG) is exposed and close to the large deletions in CTFV RdRp fragment which shortens the -helix encompassed by the 4HDG positions 636-644.
The CTFV described in this phylogenetic and evolutionary analysis have been isolated from mosquitoes collected in Portugal during the summertime (2009-2010), so as from mosquitoes collected between 2001 and 2005 in the southeast and northeast of Spain.Turkey and Myanmar had virus isolation in 2014 and 2015. This was not unexpected because Spain and Portugal are neighbor countries in geological barriers between them, and in Turkey maybe for trading, between these three countries, as previously suggested[38,39], indicating that this virus has a wider distribution and can be not restricted to a single mosquito genus or species. The different sites under selective pressure, underlining the different environmental pressure on the mosquito vectors living differently in Turkey and Iberian Peninsula, this pressure can be important, in causing a change of the vector for the virus as happened for other flaviviruses so as Chikungunya virus[28].
It has been found evidence of consistent commercial trades between Portugal and Turkey due to the development of trade agreements, these bilateral trades have started in 2014 that is the year of the first Cx. theileri flavivirus outbreak, moreover among the goods exported from Portugal to Turkey there are several potential hidings for mosquitoes (such as cables, cabinets, motor cars) and this is probably the way of spreading of Cx. theileri from Portugal to Turkey. Similarly there is a bilateral Economic and Commercial Relations between Turkey and Myanmar suggesting the possible way of virus spreading[39].
Cx. theileri are also considered as competent West Nile Virus(WNV) vectors among the mosquito species distributed throughout Europe. An interaction of CFTV and WNV in the mosquito host might be possible, referring to the nucleotide mutations for selective environmental pressure. The genome evolution probably can support the hypothesis that insect-specific flavivirus may adapt to a variety of vectors switching in different host. Phylogenetic and evolutionary analysis can be an important tool for understanding the evolutionary impact of the probable contemporary existence between nonpathogenic and pathogenic flaviviruses among these vectors. The molecular epidemiological surveillance is important to improve the knowledge about the interaction between pathogenic versus non-pathogenic viruses and host vector to control the pathogenic arboviruses epizootic in Mediterranean areas.
Conflict of interest statement
We declare that we have no conflict of interest
 Asian Pacific Journal of Tropical Medicine2019年5期
Asian Pacific Journal of Tropical Medicine2019年5期
- Asian Pacific Journal of Tropical Medicine的其它文章
- Parsonage-Turner syndrome following chikungunya virus infection:A case report
- Seroprevalence of Cryptosporidium and risks of cryptosporidiosis in residents of Sothern Egypt: A cross-sectional study
- Evaluation of phytochemical properties and larvicidal activities of Cynodon dactylon, Clerodendrum viscosum, Spilanthes acmella and Terminalia chebula against Aedes aegypti
- Climate change and potential distribution of zoonotic cutaneous leishmaniasis in Central Iran: Horizon 2030 and 2050
- Antimalarial activity of a novel series of artemisinin-derived 1, 2,3-triazole dimers